

Cell Progression and Survival Functions of Enzymes Secreted in Extracellular Vesicles Associated with Breast and Prostate Cancers

 ,
,  , and
, and
Abstract
1. Introduction
2. Roles of Enzymes in the Pathogenesis of Breast and Prostate Cancers
3. Cell Progression and Survival Functions of Enzymes Secreted in EVs Associated with Breast and Prostate Cancers
3.1. Enzymes Secreted in EVs Associated with Both Breast and Prostate Cancers
3.1.1. Adenosine Triphosphate (ATP) Citrate Lyase (ACLY)
3.1.2. Enolase (ENO)
3.1.3. Fatty Acid Synthase (FASN)
3.1.4. Focal Adhesion Kinase (FAK)
3.1.5. Pyruvate Kinase (PK)
3.2. Enzymes Secreted in EVs Associated with Breast Cancer (with No Current Evidence in Prostate Cancer)
3.2.1. Phosphoglycerate Kinase 1 (PGK1)
3.2.2. Phosphoglycerate Mutase 1 (PGAM1)
3.2.3. Glucose-6-Phosphate Dehydrogenase (G6PDH)
3.2.4. Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH)
3.2.5. A Disintegrin and Metalloproteinases (ADAM) 9 and 10
3.2.6. Matrix Metalloproteinase 1 (MMP1)
3.2.7. Peroxiredoxin (PRDX) 1 and 2
3.2.8. Sirtuin 1 and 6 (SIRT1 and SIRT6)
3.2.9. Ras Homolog A and C (RhoA and RhoC) GTPases
3.2.10. Indoleamine-2,3-Dioxygenase (IDO)
3.2.11. Tissue Transglutaminase (TTG)
3.3. Enzymes Secreted in EVs Associated with Prostate Cancer (with No Current Evidence in Breast Cancer)
3.3.1. Src Kinase (SK)
3.3.2. AKT1 Kinase
3.3.3. TMPRSS2 Serine Protease
3.3.4. Transglutaminase-4 (TGM4)
3.3.5. Six-Transmembrane Epithelial Antigen of Prostate (STEAP) 1 and 2
3.3.6. Hyaluronidase 1 (HYAL1)
4. Clinical Potential of EVs in Breast and Prostate Cancers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robinson, P.K. Enzymes: Principles and biotechnological applications. Essays Biochem. 2015, 59, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wei, M.; Jia, M.; Cao, M. Involvement of Various Enzymes in the Physiology and Pathogenesis of Streptococcus suis. Vet. Sci. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.G.N.d.; Thode, H.; Eslambolchi, Y.; Chopra, S.; Young, D.; Gill, S.; Devel, L.; Dufour, A. Matrix Metalloproteinases: From Molecular Mechanisms to Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 2022, 74, 714–770. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Kyriakopoulou, K.; Koutsakis, C.; Mastronikolis, S.; Karamanos, N.K. Key Matrix Remodeling Enzymes: Functions and Targeting in Cancer. Cancers 2021, 13, 1441. [Google Scholar] [CrossRef]
- Woodward, J.K.; Holen, I.; Coleman, R.E.; Buttle, D.J. The roles of proteolytic enzymes in the development of tumour-induced bone disease in breast and prostate cancer. Bone 2007, 41, 912–927. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA A Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- The Australian Institute of Health and Welfare. Cancer Data in Australia; AIHW: Canberra, Australia, 2022. [Google Scholar]
- Risbridger, G.P.; Davis, I.D.; Birrell, S.N.; Tilley, W.D. Breast and prostate cancer: More similar than different. Nat. Rev. Cancer 2010, 10, 205–212. [Google Scholar] [CrossRef]
- Michael, P.; Roversi, G.; Brown, K.; Sharifi, N. Adrenal Steroids and Resistance to Hormonal Blockade of Prostate and Breast Cancer. Endocrinology 2023, 164, bqac218. [Google Scholar] [CrossRef]
- Hemminki, K.; Försti, A.; Chen, B. Breast and prostate cancer: Familial associations. Nat. Rev. Cancer 2010, 10, 523. [Google Scholar] [CrossRef]
- Chen, Y.; Sadasivan, S.M.; She, R.; Datta, I.; Taneja, K.; Chitale, D.; Gupta, N.; Davis, M.B.; Newman, L.A.; Rogers, C.G.; et al. Breast and prostate cancers harbor common somatic copy number alterations that consistently differ by race and are associated with survival. BMC Med. Genom. 2020, 13, 116. [Google Scholar] [CrossRef]
- Spratt, D.E.; Jagsi, R. Breast and Prostate Cancer: Lessons to Be Shared. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Lange, P.H. Chapter 19—Breast and Prostate Cancers: A Comparison of Two Endocrinologic Malignancies. In Prostate Cancer, 2nd ed.; Mydlo, J.H., Godec, C.J., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 157–165. [Google Scholar] [CrossRef]
- Chitti, S.V.; Gummadi, S.; Kang, T.; Shahi, S.; Marzan, A.L.; Nedeva, C.; Sanwlani, R.; Bramich, K.; Stewart, S.; Petrovska, M.; et al. Vesiclepedia 2024: An extracellular vesicles and extracellular particles repository. Nucleic Acids Res. 2024, 52, D1694–D1698. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Shin, K.J.; Chae, Y.C. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp. Mol. Med. 2024, 56, 877–889. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, Q.; Jiang, L. Current Knowledge on Exosome Biogenesis, Cargo-Sorting Mechanism and Therapeutic Implications. Membranes 2022, 12, 498. [Google Scholar] [CrossRef]
- Yue, M.; Hu, S.; Sun, H.; Tuo, B.; Jia, B.; Chen, C.; Wang, W.; Liu, J.; Liu, Y.; Sun, Z.; et al. Extracellular vesicles remodel tumor environment for cancer immunotherapy. Mol. Cancer 2023, 22, 203. [Google Scholar] [CrossRef]
- Hennipman, A.; Smits, J.; van Oirschot, B.; van Houwelingen, J.C.; Rijksen, G.; Neyt, J.P.; van Unnik, J.A.M.; Staal, G.E.J. Glycolytic Enzymes in Breast Cancer, Benign Breast Disease and Normal Breast Tissue. Tumor Biol. 2009, 8, 251–263. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Li, X.; Peng, S.; Li, J.; Yan, W.; Cui, Y.; Xiao, H.; Wen, X. Increased expression of glycolytic enzymes in prostate cancer tissues and association with Gleason scores. Int. J. Clin. Exp. Pathol. 2017, 10, 11080–11089. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Kim, S.K.; Jung, W.H.; Koo, J.S. Differential Expression of Enzymes Associated with Serine/Glycine Metabolism in Different Breast Cancer Subtypes. PLoS ONE 2014, 9, e101004. [Google Scholar] [CrossRef]
- Plymate, S.R.; Sprenger, C.; Haffner, M.C. Starving lethal prostate cancer by targeting heat shock proteins and glycolytic enzymes. Cell Rep. Med. 2022, 3, 100493. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ma, H.; Wang, J.; Huang, M.; Fu, D.; Qin, L.; Yin, Q. Energy metabolism pathways in breast cancer progression: The reprogramming, crosstalk, and potential therapeutic targets. Transl. Oncol. 2022, 26, 101534. [Google Scholar] [CrossRef]
- Starlard-Davenport, A.; Lyn-Cook, B.; Radominska-Pandya, A. Novel identification of UDP-glucuronosyltransferase 1A10 as an estrogen-regulated target gene. Steroids 2008, 73, 139–147. [Google Scholar] [CrossRef]
- Li, Y.; Steppi, A.; Zhou, Y.; Mao, F.; Miller, P.C.; He, M.M.; Zhao, T.; Sun, Q.; Zhang, J. Tumoral expression of drug and xenobiotic metabolizing enzymes in breast cancer patients of different ethnicities with implications to personalized medicine. Sci. Rep. 2017, 7, 4747. [Google Scholar] [CrossRef]
- Chen, T.C.; Sakaki, T.; Yamamoto, K.; Kittaka, A. The Roles of Cytochrome P450 Enzymes in Prostate Cancer Development and Treatment. Anticancer. Res. 2012, 32, 291–298. [Google Scholar]
- Honma, N.; Takubo, K.; Sawabe, M.; Arai, T.; Akiyama, F.; Sakamoto, G.; Utsumi, T.; Yoshimura, N.; Harada, N. Estrogen-Metabolizing Enzymes in Breast Cancers from Women over the Age of 80 Years. J. Clin. Endocrinol. Metab. 2006, 91, 607–613. [Google Scholar] [CrossRef]
- Pasqualini, J.R.; Chetrite, G.S. Recent insight on the control of enzymes involved in estrogen formation and transformation in human breast cancer. J. Steroid Biochem. Mol. Biol. 2005, 93, 221–236. [Google Scholar] [CrossRef]
- Pasqualini, J.R. Breast cancer and steroid metabolizing enzymes: The role of progestogens. Maturitas 2009, 65 (Suppl. S1), S17–S21. [Google Scholar] [CrossRef]
- Myutan, K.; Mohamed, S.; Kefah, M. Oestrogen-Synthesising Enzymes and Breast Cancer. Anticancer. Res. 2009, 29, 1095. [Google Scholar]
- Suzuki, T.; Miki, Y.; Nakamura, Y.; Moriya, T.; Ito, K.; Ohuchi, N.; Sasano, H. Sex steroid-producing enzymes in human breast cancer. Endocr. Relat. Cancer 2005, 12, 701–720. [Google Scholar] [CrossRef] [PubMed]
- Soronen, P.; Laiti, M.; Törn, S.; Härkönen, P.; Patrikainen, L.; Li, Y.; Pulkka, A.; Kurkela, R.; Herrala, A.; Kaija, H.; et al. Sex steroid hormone metabolism and prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Suzuki, T.; Nakabayashi, M.; Endoh, M.; Sakamoto, K.; Mikami, Y.; Moriya, T.; Ito, A.; Takahashi, S.; Yamada, S.; et al. In situ androgen producing enzymes in human prostate cancer. Endocr. Relat. Cancer 2005, 12, 101–107. [Google Scholar] [CrossRef]
- McNamara, K.M.; Sasano, H. The role of 17βHSDs in breast tissue and breast cancers. Mol. Cell. Endocrinol. 2019, 489, 32–44. [Google Scholar] [CrossRef]
- Mindnich, R.; Möller, G.; Adamski, J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2004, 218, 7–20. [Google Scholar] [CrossRef]
- Collin, L.J.; Ulrichsen, S.P.; Ahern, T.P.; Goodman, M.; McCullough, L.E.; Waller, L.A.; Bang Christensen, K.; Damkier, P.; Hamilton-Dutoit, S.; Lauridsen, K.L.; et al. 17β-Hydroxysteroid dehydrogenase 1:2 and breast cancer recurrence: A Danish population-based study. Acta Oncol. 2020, 59, 329–333. [Google Scholar] [CrossRef]
- Hilborn, E.; Stål, O.; Jansson, A. Estrogen and androgen-converting enzymes 17β-hydroxysteroid dehydrogenase and their involvement in cancer: With a special focus on 17β-hydroxysteroid dehydrogenase type 1, 2, and breast cancer. Oncotarget 2017, 8, 30552. [Google Scholar]
- Ning, X.; Yang, Y.; Deng, H.; Zhang, Q.; Huang, Y.; Su, Z.; Fu, Y.; Xiang, Q.; Zhang, S. Development of 17β-hydroxysteroid dehydrogenase type 3 as a target in hormone-dependent prostate cancer therapy. Steroids 2017, 121, 10–16. [Google Scholar] [CrossRef]
- Denu, J.M. The Sir 2 family of protein deacetylases. Curr. Opin. Chem. Biol. 2005, 9, 431–440. [Google Scholar] [CrossRef]
- Vassilopoulos, A.; Fritz, K.S.; Petersen, D.R.; Gius, D. The human sirtuin family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Onyiba, C.I.; Scarlett, C.J.; Weidenhofer, J. The Mechanistic Roles of Sirtuins in Breast and Prostate Cancer. Cancers 2022, 14, 5118. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gai, J.W.; Wang, Y.; Jin, H.F.; Du, J.B.; Jin, J. Characterization of hydrogen sulfide and its synthases, cystathionine β-synthase and cystathionine γ-lyase, in human prostatic tissue and cells. Urology 2012, 79, 483.e481–485. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Roy-Chowdhuri, S.; Bose, S.; Yoon, A.; Faull, K.; Farias-Eisner, R.; et al. Role of cystathionine β-synthase in human breast Cancer. Free Radic. Biol. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef]
- Nafea, H.; Youness, R.A.; Dawoud, A.; Khater, N.; Manie, T.; Abdel-Kader, R.; Bourquin, C.; Szabo, C.; Gad, M.Z. Dual targeting of H(2)S synthesizing enzymes; cystathionine β-synthase and cystathionine γ-lyase by miR-939-5p effectively curbs triple negative breast cancer. Heliyon 2023, 9, e21063. [Google Scholar] [CrossRef]
- Khattak, S.; Rauf, M.A.; Khan, N.H.; Zhang, Q.-Q.; Chen, H.-J.; Muhammad, P.; Ansari, M.A.; Alomary, M.N.; Jahangir, M.; Zhang, C.-Y.; et al. Hydrogen Sulfide Biology and Its Role in Cancer. Molecules 2022, 27, 3389. [Google Scholar] [CrossRef]
- Siiskonen, H.; Oikari, S.; Pasonen-Seppänen, S.; Rilla, K. Hyaluronan synthase 1: A mysterious enzyme with unexpected functions. Front. Immunol. 2015, 6, 43. [Google Scholar] [CrossRef]
- Auvinen, P.; Rilla, K.; Tumelius, R.; Tammi, M.; Sironen, R.; Soini, Y.; Kosma, V.M.; Mannermaa, A.; Viikari, J.; Tammi, R. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res. Treat. 2014, 143, 277–286. [Google Scholar] [CrossRef]
- Li, P.; Xiang, T.; Li, H.; Li, Q.; Yang, B.; Huang, J.; Zhang, X.; Shi, Y.; Tan, J.; Ren, G. Hyaluronan synthase 2 overexpression is correlated with the tumorigenesis and metastasis of human breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12101–12114. [Google Scholar]
- Karousou, E.; Parnigoni, A.; Moretto, P.; Passi, A.; Viola, M.; Vigetti, D. Hyaluronan in the Cancer Cells Microenvironment. Cancers 2023, 15, 798. [Google Scholar] [CrossRef]
- Simpson, M.A. Concurrent expression of hyaluronan biosynthetic and processing enzymes promotes growth and vascularization of prostate tumors in mice. Am. J. Pathol. 2006, 169, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Gao, F.; Han, Z.; Xu, X.; Underhill, C.B.; Zhang, L. Hyaluronan synthase 3 overexpression promotes the growth of TSU prostate cancer cells. Cancer Res. 2001, 61, 5207–5214. [Google Scholar] [PubMed]
- Velesiotis, C.; Vasileiou, S.; Vynios, D.H. A guide to hyaluronan and related enzymes in breast cancer: Biological significance and diagnostic value. FEBS J. 2019, 286, 3057–3074. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, P.; Maddela, J.J.; Mani, S.A. Spatial and Temporal Relationship between Epithelial–Mesenchymal Transition (EMT) and Stem Cells in Cancer. Clin. Chem. 2024, 70, 190–205. [Google Scholar] [CrossRef]
- Karalis, T. Targeting Hyaluronan Synthesis in Cancer: A Road Less Travelled. Biologics 2023, 3, 402–414. [Google Scholar] [CrossRef]
- Eatemadi, A.; Aiyelabegan, H.T.; Negahdari, B.; Mazlomi, M.A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed. Pharmacother. 2017, 86, 221–231. [Google Scholar] [CrossRef]
- Tagirasa, R.; Yoo, E. Role of Serine Proteases at the Tumor-Stroma Interface. Front. Immunol. 2022, 13, 832418. [Google Scholar] [CrossRef]
- Kim, S. TMPRSS4, a type II transmembrane serine protease, as a potential therapeutic target in cancer. Exp. Mol. Med. 2023, 55, 716–724. [Google Scholar] [CrossRef]
- Navasatli, S.A.; Vahdati, S.N.; Arjmand, T.F.; Behboudi, H. New insight into the role of the ADAM protease family in breast carcinoma progression. Heliyon 2024, 10, e24805. [Google Scholar] [CrossRef]
- Koistinen, H.; Kovanen, R.-M.; Hollenberg, M.D.; Dufour, A.; Radisky, E.S.; Stenman, U.-H.; Batra, J.; Clements, J.; Hooper, J.D.; Diamandis, E.; et al. The roles of proteases in prostate cancer. IUBMB Life 2023, 75, 493–513. [Google Scholar] [CrossRef]
- Park, K.C.; Dharmasivam, M.; Richardson, D.R. The Role of Extracellular Proteases in Tumor Progression and the Development of Innovative Metal Ion Chelators that Inhibit their Activity. Int. J. Mol. Sci. 2020, 21, 6805. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J. Matrix metalloproteinases as therapeutic targets in breast cancer. Front. Oncol. 2023, 12, 1108695. [Google Scholar] [CrossRef] [PubMed]
- Escaff, S.; Fernández, J.M.; González, L.O.; Suárez, A.; González-Reyes, S.; González, J.M.; Vizoso, F.J. Study of matrix metalloproteinases and their inhibitors in prostate cancer. Br. J. Cancer 2010, 102, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-H.; Liao, C.-H.; Huang, W.-C.; Chang, W.-S.; Wu, H.-C.; Hsu, S.-W.; Chen, K.-Y.; Wang, Z.-H.; Hsia, T.-C.; Bau, D.-T.; et al. Association of Matrix Metalloproteinase-2 Genotypes With Prostate Cancer Risk. Anticancer. Res. 2023, 43, 343–349. [Google Scholar] [CrossRef]
- Luo, J.; Zou, H.; Guo, Y.; Tong, T.; Ye, L.; Zhu, C.; Deng, L.; Wang, B.; Pan, Y.; Li, P. SRC kinase-mediated signaling pathways and targeted therapies in breast cancer. Breast Cancer Res. 2022, 24, 99. [Google Scholar] [CrossRef]
- García-Aranda, M.; Redondo, M. Protein Kinase Targets in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2543. [Google Scholar] [CrossRef]
- Miller, K.J.; Asim, M. Unravelling the Role of Kinases That Underpin Androgen Signalling in Prostate Cancer. Cells 2022, 11, 952. [Google Scholar] [CrossRef]
- Bagheri, S.; Rahban, M.; Bostanian, F.; Esmaeilzadeh, F.; Bagherabadi, A.; Zolghadri, S.; Stanek, A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 2022, 14, 515. [Google Scholar] [CrossRef]
- Gali, A.; Bijnsdorp, I.V.; Piersma, S.R.; Pham, T.V.; Gutiérrez-Galindo, E.; Kühnel, F.; Tsolakos, N.; Jimenez, C.R.; Hausser, A.; Alexopoulos, L.G. Protein kinase D drives the secretion of invasion mediators in triple-negative breast cancer cell lines. iScience 2024, 27, 108958. [Google Scholar] [CrossRef]
- Roy, A.; Prasad, S.; Chen, Y.; Chao, Y.; Liu, Y.; Zhao, J.; Wang, Q.J. Protein Kinase D2 and D3 Promote Prostate Cancer Cell Bone Metastasis by Positively Regulating Runx2 in a MEK/ERK1/2–Dependent Manner. Am. J. Pathol. 2023, 193, 624–637. [Google Scholar] [CrossRef]
- Durand, N.; Borges, S.; Storz, P. Functional and therapeutic significance of protein kinase D enzymes in invasive breast cancer. Cell. Mol. Life Sci. 2015, 72, 4369–4382. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Fingleton, B. Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis. Clin. Exp. Metastasis 2019, 36, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Qi, Y.; Yang, W. The Noncanonical Functions of Metabolites in Tumor Progression. Metabolites 2024, 14, 171. [Google Scholar] [CrossRef]
- Shegay, P.V.; Shatova, O.P.; Zabolotneva, A.A.; Shestopalov, A.V.; Kaprin, A.D. Moonlight functions of glycolytic enzymes in cancer. Front. Mol. Biosci. 2023, 10, 1076138. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y. Nonmetabolic functions of metabolic enzymes in cancer development. Cancer Commun. 2018, 38, 63. [Google Scholar] [CrossRef]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, Y.; Yang, W.; Han, Y.; Chen, Y.; Oyang, L.; Lin, J.; et al. Metabolic reprogramming and epigenetic modifications in cancer: From the impacts and mechanisms to the treatment potential. Exp. Mol. Med. 2023, 55, 1357–1370. [Google Scholar] [CrossRef]
- Berumen Sánchez, G.; Bunn, K.E.; Pua, H.H.; Rafat, M. Extracellular vesicles: Mediators of intercellular communication in tissue injury and disease. Cell Commun. Signal. 2021, 19, 104. [Google Scholar] [CrossRef]
- Winzer, R.; Nguyen, D.H.; Schoppmeier, F.; Cortesi, F.; Gagliani, N.; Tolosa, E. Purinergic enzymes on extracellular vesicles: Immune modulation on the go. Front. Immunol. 2024, 15, 1362996. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Shomar, A.; Barak, O.; Brenner, N. Cancer progression as a learning process. iScience 2022, 25, 103924. [Google Scholar] [CrossRef]
- Albitre, A.; Reglero, C.; González-Muñoz, T.; Penela, P. The stress connection in cancer: The adrenergic fuelling of breast tumors. Curr. Opin. Physiol. 2023, 36, 100720. [Google Scholar] [CrossRef]
- Minic, Z.; Hüttmann, N.; Poolsup, S.; Li, Y.; Susevski, V.; Zaripov, E.; Berezovski, M.V. Phosphoproteomic Analysis of Breast Cancer-Derived Small Extracellular Vesicles Reveals Disease-Specific Phosphorylated Enzymes. Biomedicines 2022, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Dalla, P.V.; Santos, J.; Milthorpe, B.K.; Padula, M.P. Selectively Packaged Proteins in Breast Cancer Extracellular Vesicles Involved in Metastasis. Int. J. Mol. Sci. 2020, 21, 4990. [Google Scholar] [CrossRef]
- Minic, Z.; Li, Y.; Hüttmann, N.; Uppal, G.K.; D’Mello, R.; Berezovski, M.V. Lysine Acetylome of Breast Cancer-Derived Small Extracellular Vesicles Reveals Specific Acetylation Patterns for Metabolic Enzymes. Biomedicines 2023, 11, 1076. [Google Scholar] [CrossRef]
- Smyth, T.J.; Redzic, J.S.; Graner, M.W.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells In Vitro. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 2954–2965. [Google Scholar] [CrossRef]
- Galindo-Hernandez, O.; Villegas-Comonfort, S.; Candanedo, F.; González-Vázquez, M.C.; Chavez-Ocaña, S.; Jimenez-Villanueva, X.; Sierra-Martinez, M.; Salazar, E.P. Elevated concentration of microvesicles isolated from peripheral blood in breast cancer patients. Arch. Med. Res. 2013, 44, 208–214. [Google Scholar] [CrossRef]
- Pallares-Rusiñol, A.; Moura, S.L.; Martí, M.; Pividori, M.I. Electrochemical Genosensing of Overexpressed GAPDH Transcripts in Breast Cancer Exosomes. Anal. Chem. 2023, 95, 2487–2495. [Google Scholar] [CrossRef]
- Green, T.M.; Alpaugh, M.L.; Barsky, S.H.; Rappa, G.; Lorico, A. Breast Cancer-Derived Extracellular Vesicles: Characterization and Contribution to the Metastatic Phenotype. Biomed. Res. Int. 2015, 2015, 634865. [Google Scholar] [CrossRef]
- Risha, Y.; Minic, Z.; Ghobadloo, S.M.; Berezovski, M.V. The proteomic analysis of breast cell line exosomes reveals disease patterns and potential biomarkers. Sci. Rep. 2020, 10, 13572. [Google Scholar] [CrossRef]
- Rontogianni, S.; Synadaki, E.; Li, B.; Liefaard, M.C.; Lips, E.H.; Wesseling, J.; Wu, W.; Altelaar, M. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Commun. Biol. 2019, 2, 325. [Google Scholar] [CrossRef]
- Ando, W.; Kikuchi, K.; Uematsu, T.; Yokomori, H.; Takaki, T.; Sogabe, M.; Kohgo, Y.; Otori, K.; Ishikawa, S.; Okazaki, I. Novel breast cancer screening: Combined expression of miR-21 and MMP-1 in urinary exosomes detects 95% of breast cancer without metastasis. Sci. Rep. 2019, 9, 13595. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tao, Z.; Chen, Y.; Lin, S.; Zhu, M.; Ji, W.; Liu, X.; Li, T.; Hu, X. Exosomal MMP-1 transfers metastasis potential in triple-negative breast cancer through PAR1-mediated EMT. Breast Cancer Res. Treat. 2022, 193, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, G.; Albanese, N.N.; Di Cara, G.; Gygax, D.; Vittorelli, M.L.; Pucci-Minafra, I. Proteomic Analysis of Exosome-like Vesicles Derived from Breast Cancer Cells. Anticancer. Res. 2012, 32, 847–860. [Google Scholar]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol. Biol. Cell 2014, 25, 17–29. [Google Scholar] [CrossRef]
- Isla Larrain, M.T.; Rabassa, M.E.; Lacunza, E.; Barbera, A.; Cretón, A.; Segal-Eiras, A.; Croce, M.V. IDO is highly expressed in breast cancer and breast cancer-derived circulating microvesicles and associated to aggressive types of tumors by in silico analysis. Tumor Biol. 2014, 35, 6511–6519. [Google Scholar] [CrossRef]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef]
- Hosseini-Beheshti, E.; Pham, S.; Adomat, H.; Li, N.; Tomlinson Guns, E.S. Exosomes as biomarker enriched microvesicles: Characterization of exosomal proteins derived from a panel of prostate cell lines with distinct AR phenotypes. Mol. Cell Proteom. 2012, 11, 863–885. [Google Scholar] [CrossRef]
- DeRita, R.M.; Zerlanko, B.; Singh, A.; Lu, H.; Iozzo, R.V.; Benovic, J.L.; Languino, L.R. c-Src, Insulin-Like Growth Factor I Receptor, G-Protein-Coupled Receptor Kinases and Focal Adhesion Kinase are Enriched Into Prostate Cancer Cell Exosomes. J. Cell Biochem. 2017, 118, 66–73. [Google Scholar] [CrossRef]
- Dai, J.; Escara-Wilke, J.; Keller, J.M.; Jung, Y.; Taichman, R.S.; Pienta, K.J.; Keller, E.T. Primary prostate cancer educates bone stroma through exosomal pyruvate kinase M2 to promote bone metastasis. J. Exp. Med. 2019, 216, 2883–2899. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Spinelli, C.; Reis-Sobreiro, M.; Cavallini, L.; You, S.; Zandian, M.; Li, X.; Mishra, R.; Chiarugi, P.; Adam, R.M.; et al. MYC Mediates Large Oncosome-Induced Fibroblast Reprogramming in Prostate Cancer. Cancer Res. 2017, 77, 2306–2317. [Google Scholar] [CrossRef]
- Lázaro-Ibáñez, E.; Lunavat, T.R.; Jang, S.C.; Escobedo-Lucea, C.; Oliver-De La Cruz, J.; Siljander, P.; Lötvall, J.; Yliperttula, M. Distinct prostate cancer-related mRNA cargo in extracellular vesicle subsets from prostate cell lines. BMC Cancer 2017, 17, 92. [Google Scholar] [CrossRef] [PubMed]
- Sequeiros, T.; Rigau, M.; Chiva, C.; Montes, M.; Garcia-Grau, I.; Garcia, M.; Diaz, S.; Celma, A.; Bijnsdorp, I.; Campos, A.; et al. Targeted proteomics in urinary extracellular vesicles identifies biomarkers for diagnosis and prognosis of prostate cancer. Oncotarget 2017, 8, 4960–4976. [Google Scholar] [CrossRef]
- Casanova-Salas, I.; Aguilar, D.; Cordoba-Terreros, S.; Agundez, L.; Brandariz, J.; Herranz, N.; Mas, A.; Gonzalez, M.; Morales-Barrera, R.; Sierra, A.; et al. Circulating tumor extracellular vesicles to monitor metastatic prostate cancer genomics and transcriptomic evolution. Cancer Cell 2024, 42, 1301–1312.e1307. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Mizzoni, D.; Capasso, C.; Del Prete, S.; Di Raimo, R.; Falchi, M.; Angelini, D.F.; Sciarra, A.; Maggi, M.; Supuran, C.T.; et al. Plasmatic exosomes from prostate cancer patients show increased carbonic anhydrase IX expression and activity and low pH. J. Enzym. Inhib. Med. Chem. 2020, 35, 280–288. [Google Scholar] [CrossRef]
- Khanna, K.; Salmond, N.; Lynn, K.S.; Leong, H.S.; Williams, K.C. Clinical significance of STEAP1 extracellular vesicles in prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 802–811. [Google Scholar] [CrossRef]
- McAtee, C.O.; Booth, C.; Elowsky, C.; Zhao, L.; Payne, J.; Fangman, T.; Caplan, S.; Henry, M.D.; Simpson, M.A. Prostate tumor cell exosomes containing hyaluronidase Hyal1 stimulate prostate stromal cell motility by engagement of FAK-mediated integrin signaling. Matrix Biol. 2019, 78, 165–179. [Google Scholar] [CrossRef]
- McAtee, C.O.; Berkebile, A.R.; Elowsky, C.G.; Fangman, T.; Barycki, J.J.; Wahl, J.K.; Khalimonchuk, O.; Naslavsky, N.; Caplan, S.; Simpson, M.A. Hyaluronidase Hyal1 Increases Tumor Cell Proliferation and Motility through Accelerated Vesicle Trafficking. J. Biol. Chem. 2015, 290, 13144–13156. [Google Scholar] [CrossRef]
- Wang, D.; Yin, L.; Wei, J.; Yang, Z.; Jiang, G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumor Biol. 2017, 39, 1010428317698338. [Google Scholar] [CrossRef]
- Shah, S.; Carriveau, W.J.; Li, J.; Campbell, S.L.; Kopinski, P.K.; Lim, H.W.; Daurio, N.; Trefely, S.; Won, K.J.; Wallace, D.C.; et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016, 7, 43713–43730. [Google Scholar] [CrossRef]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: Evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef]
- Velez, B.C.; Petrella, C.P.; DiSalvo, K.H.; Cheng, K.; Kravtsov, R.; Krasniqi, D.; Krucher, N.A. Combined inhibition of ACLY and CDK4/6 reduces cancer cell growth and invasion. Oncol. Rep. 2023, 49, 32. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhou, Q.; Zhao, C.; Li, J.; Yu, Z.; Zhu, Q. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce hepatocellular carcinoma cell apoptosis via p-eIF2α/ATF4/CHOP axis. J. Cell Mol. Med. 2021, 25, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Islam, M.S.; Tian, J.; Lui, V.W.Y.; Xiao, D. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 2014, 349, 15–25. [Google Scholar] [CrossRef]
- Migita, T.; Okabe, S.; Ikeda, K.; Igarashi, S.; Sugawara, S.; Tomida, A.; Taguchi, R.; Soga, T.; Seimiya, H. Inhibition of ATP Citrate Lyase Induces an Anticancer Effect via Reactive Oxygen Species: AMPK as a Predictive Biomarker for Therapeutic Impact. Am. J. Pathol. 2013, 182, 1800–1810. [Google Scholar] [CrossRef]
- Zaidi, N.; Royaux, I.; Swinnen, J.V.; Smans, K. ATP Citrate Lyase Knockdown Induces Growth Arrest and Apoptosis through Different Cell- and Environment-Dependent Mechanisms. Mol. Cancer Ther. 2012, 11, 1925–1935. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Ding, M.; Zhang, H.; Xu, X.; Tang, J. Molecular mechanisms and clinical applications of miR-22 in regulating malignant progression in human cancer (Review). Int. J. Oncol. 2017, 50, 345–355. [Google Scholar] [CrossRef]
- Tu, S.-H.; Chang, C.-C.; Chen, C.-S.; Tam, K.-W.; Wang, Y.-J.; Lee, C.-H.; Lin, H.-W.; Cheng, T.-C.; Huang, C.-S.; Chu, J.-S.; et al. Increased expression of enolase α in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 539–553. [Google Scholar] [CrossRef]
- Cancemi, P.; Buttacavoli, M.; Roz, E.; Feo, S. Expression of Alpha-Enolase (ENO1), Myc Promoter-Binding Protein-1 (MBP-1) and Matrix Metalloproteinases (MMP-2 and MMP-9) Reflect the Nature and Aggressiveness of Breast Tumors. Int. J. Mol. Sci. 2019, 20, 3952. [Google Scholar] [CrossRef]
- Santiago, K.R.; Duran, A.M.; Casiano, C.A.; Almaguel, F.G.; Das, B. Abstract C063: Enolase-1 as a candidate theranostics target for neuroendocrine prostate cancer. Cancer Epidemiol. Biomark. Prev. 2023, 32, C063. [Google Scholar] [CrossRef]
- Zhou, Y.; Zeng, F.; Richards, G.O.; Wang, N. ENO2, a Glycolytic Enzyme, Contributes to Prostate Cancer Metastasis: A Systematic Review of Literature. Cancers 2024, 16, 2503. [Google Scholar] [CrossRef]
- Jin, Q.; Yuan, L.X.; Boulbes, D.; Baek, J.M.; Wang, Y.N.; Gomez-Cabello, D.; Hawke, D.H.; Yeung, S.C.; Lee, M.H.; Hortobagyi, G.N.; et al. Fatty acid synthase phosphorylation: A novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010, 12, R96. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Yuan, T.-T.; Chuang, C.-F.; Huang, Y.-T.; Chung, I.-C.; Huang, W.-C. A Novel Enolase-1 Antibody Targets Multiple Interacting Players in the Tumor Microenvironment of Advanced Prostate Cancer. Mol. Cancer Ther. 2022, 21, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, T.; Dong, L.; Li, T.; Xue, H.; Gao, B.; Ding, X.; Wang, H.; Li, H. Fatty acid synthase promotes breast cancer metastasis by mediating changes in fatty acid metabolism. Oncol. Lett. 2021, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat. Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R.; Colomer, R. Inhibition of Tumor-associated Fatty Acid Synthase Hyperactivity Induces Synergistic Chemosensitization of HER-2/neu-Overexpressing Human Breast Cancer Cells to Docetaxel (taxotere). Breast Cancer Res. Treat. 2004, 84, 183–195. [Google Scholar] [CrossRef]
- Al-Bahlani, S.; Al-Lawati, H.; Al-Adawi, M.; Al-Abri, N.; Al-Dhahli, B.; Al-Adawi, K. Fatty acid synthase regulates the chemosensitivity of breast cancer cells to cisplatin-induced apoptosis. Apoptosis 2017, 22, 865–876. [Google Scholar] [CrossRef]
- Schroeder, B.; Vander Steen, T.; Espinoza, I.; Venkatapoorna, C.M.K.; Hu, Z.; Silva, F.M.; Regan, K.; Cuyàs, E.; Meng, X.W.; Verdura, S.; et al. Fatty acid synthase (FASN) regulates the mitochondrial priming of cancer cells. Cell Death Dis. 2021, 12, 977. [Google Scholar] [CrossRef]
- Rossi, S.; Graner, E.; Febbo, P.; Weinstein, L.; Bhattacharya, N.; Onody, T.; Bubley, G.; Balk, S.; Loda, M. Fatty Acid Synthase Expression Defines Distinct Molecular Signatures in Prostate Cancer1. Mol. Cancer Res. 2003, 1, 707–715. [Google Scholar]
- Shah, U.S.; Dhir, R.; Gollin, S.M.; Chandran, U.R.; Lewis, D.; Acquafondata, M.; Pflug, B.R. Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma. Human. Pathol. 2006, 37, 401–409. [Google Scholar] [CrossRef]
- Oh, J.E.; Jung, B.H.; Park, J.; Kang, S.; Lee, H. Deciphering Fatty Acid Synthase Inhibition-Triggered Metabolic Flexibility in Prostate Cancer Cells through Untargeted Metabolomics. Cells 2020, 9, 2447. [Google Scholar] [CrossRef]
- Sadowski, M.C.; Pouwer, R.H.; Gunter, J.H.; Lubik, A.A.; Quinn, R.J.; Nelson, C.C. The fatty acid synthase inhibitor triclosan: Repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget 2014, 5, 9362. [Google Scholar] [PubMed]
- De Schrijver, E.; Brusselmans, K.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Silencing of the Fatty Acid Synthase Gene by RNA Interference Inhibits Growth and Induces Apoptosis of LNCaP Prostate Cancer Cells. In Hormonal Carcinogenesis IV; Li, J.J., Li, S.A., Llombart-Bosch, A., Eds.; Springer US: Boston, MA, USA, 2005; pp. 350–356. [Google Scholar] [CrossRef]
- Rae, C.; Haberkorn, U.; Babich, J.W.; Mairs, R.J. Inhibition of Fatty Acid Synthase Sensitizes Prostate Cancer Cells to Radiotherapy. Radiat. Res. 2015, 184, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Koizumi, A.; Narita, S.; Inoue, T.; Tsuchiya, N.; Nakanishi, H.; Numakura, K.; Tsuruta, H.; Saito, M.; Satoh, S.; et al. Diet-induced alteration of fatty acid synthase in prostate cancer progression. Oncogenesis 2016, 5, e195. [Google Scholar] [CrossRef]
- Li, J.; Dong, L.; Wei, D.; Wang, X.; Zhang, S.; Li, H. Fatty Acid Synthase Mediates the Epithelial-Mesenchymal Transition of Breast Cancer Cells. Int. J. Biol. Sci. 2014, 10, 171–180. [Google Scholar] [CrossRef]
- Serhan, H.A.; Bao, L.; Cheng, X.; Qin, Z.; Liu, C.-J.; Heth, J.A.; Udager, A.M.; Soellner, M.B.; Merajver, S.D.; Morikawa, A.; et al. Targeting fatty acid synthase in preclinical models of TNBC brain metastases synergizes with SN-38 and impairs invasion. NPJ Breast Cancer 2024, 10, 43. [Google Scholar] [CrossRef]
- Yoshii, Y.; Furukawa, T.; Oyama, N.; Hasegawa, Y.; Kiyono, Y.; Nishii, R.; Waki, A.; Tsuji, A.B.; Sogawa, C.; Wakizaka, H.; et al. Fatty Acid Synthase Is a Key Target in Multiple Essential Tumor Functions of Prostate Cancer: Uptake of Radiolabeled Acetate as a Predictor of the Targeted Therapy Outcome. PLoS ONE 2013, 8, e64570. [Google Scholar] [CrossRef]
- De Piano, M.; Manuelli, V.; Zadra, G.; Loda, M.; Muir, G.; Chandra, A.; Morris, J.; Van Hemelrijck, M.; Wells, C.M. Exploring a role for fatty acid synthase in prostate cancer cell migration. Small GTPases 2021, 12, 265–272. [Google Scholar] [CrossRef]
- Ganguly, K.K.; Sen, T.; Mandal, S.; Biswas, J.; Chatterjee, A. Studies on Focal Adhesion Kinase in Human Breast Cancer Tissue. J. Cancer Ther. 2012, 3, 7–19. [Google Scholar]
- Rovin, J.D.; Frierson, H.F., Jr.; Ledinh, W.; Parsons, J.T.; Adams, R.B. Expression of focal adhesion kinase in normal and pathologic human prostate tissues. Prostate 2002, 53, 124–132. [Google Scholar] [CrossRef]
- Xu, L.-H.; Yang, X.; Bradham, C.A.; Brenner, D.A.; Baldwin, A.S., Jr.; Craven, R.J.; Cance, W.G. The Focal Adhesion Kinase Suppresses Transformation-associated, Anchorage-independent Apoptosis in Human Breast Cancer Cells: Involvement of Death Receptor-Related Signaling Pathways. J. Biol. Chem. 2000, 275, 30597–30604. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Beviglia, L.; Xu, L.-H.; Earp, H.S., III; Craven, R.; Cance, W. Dual Inhibition of Focal Adhesion Kinase and Epidermal Growth Factor Receptor Pathways Cooperatively Induces Death Receptor-mediated Apoptosis in Human Breast Cancer Cells. J. Biol. Chem. 2002, 277, 38978–38987. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.H.; Surmacz, T.A.; Nyormoi, O.; Whitacre, C.M. Inhibition of focal adhesion kinase by antisense oligonucleotides enhances the sensitivity of breast cancer cells to camptothecins. Biocell 2003, 27, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Danta, C.C.; Sahu, A.N. Chapter 16—Naturally occurring anticancer drugs. In Medicinal Chemistry of Chemotherapeutic Agents; Acharya, P.C., Kurosu, M., Eds.; Academic Press: San Diego, CA, USA, 2023; pp. 539–588. [Google Scholar] [CrossRef]
- Lazaro, G.; Smith, C.; Goddard, L.; Jordan, N.; McClelland, R.; Barrett-Lee, P.; Nicholson, R.I.; Hiscox, S. Targeting focal adhesion kinase in ER+/HER2+ breast cancer improves trastuzumab response. Endocr. Relat. Cancer 2013, 20, 691–704. [Google Scholar] [CrossRef]
- Rovin, J.D.; Ledinh, W.; Parsons, T.J.; Adams, R.B. Inhibition of focal adhesion kinase prevents growth of prostate tumors in mice. J. Am. Coll. Surg. 2000, 191, S87. [Google Scholar] [CrossRef]
- Lin, H.-M.; Lee, B.Y.; Castillo, L.; Spielman, C.; Grogan, J.; Yeung, N.K.; Kench, J.G.; Stricker, P.D.; Haynes, A.-M.; Centenera, M.M.; et al. Effect of FAK inhibitor VS-6063 (defactinib) on docetaxel efficacy in prostate cancer. Prostate 2018, 78, 308–317. [Google Scholar] [CrossRef]
- Johnson, T.R.; Khandrika, L.; Kumar, B.; Venezia, S.; Koul, S.; Chandhoke, R.; Maroni, P.; Donohue, R.; Meacham, R.B.; Koul, H.K. Focal adhesion kinase controls aggressive phenotype of androgen-independent prostate cancer. Mol. Cancer Res. 2008, 6, 1639–1648. [Google Scholar] [CrossRef]
- Hiscox, S.; Barnfather, P.; Hayes, E.; Bramble, P.; Christensen, J.; Nicholson, R.I.; Barrett-Lee, P. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res. Treat. 2011, 125, 659–669. [Google Scholar] [CrossRef]
- Sumitomo, M.; Shen, R.; Walburg, M.; Dai, J.; Geng, Y.; Navarro, D.; Boileau, G.; Papandreou, C.N.; Giancotti, F.G.; Knudsen, B.; et al. Neutral endopeptidase inhibits prostate cancer cell migration by blocking focal adhesion kinase signaling. J. Clin. Investig. 2000, 106, 1399–1407. [Google Scholar] [CrossRef]
- Albrecht, M.; Gillen, S.; Wilhelm, B.; Doroszewicz, J.; Aumüller, G. Expression, Localization and Activity of Neutral Endopeptidase in Cultured Cells of Benign Prostatic Hyperplasia and Prostate Cancer. J. Urol. 2002, 168, 336–342. [Google Scholar] [CrossRef]
- Lacoste, J.; Aprikian, A.G.; Chevalier, S. Focal adhesion kinase is required for bombesin-induced prostate cancer cell motility. Mol. Cell. Endocrinol. 2005, 235, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, T.; Varkaris, A.S.; Parikh, N.U.; Song, J.H.; Cheng, C.-J.; Schweppe, R.E.; Alexander, S.; Davis, J.W.; Troncoso, P.; Friedl, P.; et al. Yes-mediated phosphorylation of focal adhesion kinase at tyrosine 861 increases metastatic potential of prostate cancer cells. Oncotarget 2015, 6, 10175. [Google Scholar] [PubMed]
- Chen, W.; Wang, J. RAB11A Promotes Cell Malignant Progression and Tumor Formation of Prostate Cancer via Activating FAK/AKT Signaling Pathway. Evid.-Based Complement. Altern. Med. 2023, 2023, 5885387. [Google Scholar] [CrossRef]
- Lv, Z.; Li, W.; Wei, X. S100A9 promotes prostate cancer cell invasion by activating TLR4/NF-κB/integrin β1/FAK signaling. OncoTargets Ther. 2020, 13, 6443–6452. [Google Scholar] [CrossRef]
- Ma, C.; Zu, X.; Liu, K.; Bode, A.M.; Dong, Z.; Liu, Z.; Kim, D.J. Knockdown of Pyruvate Kinase M Inhibits Cell Growth and Migration by Reducing NF-kB Activity in Triple-Negative Breast Cancer Cells. Mol. Cells 2019, 42, 628–636. [Google Scholar] [CrossRef]
- Jiang, K.; He, B.; Lai, L.; Chen, Q.; Liu, Y.; Guo, Q.; Wang, Q. Cyclosporine A inhibits breast cancer cell growth by downregulating the expression of pyruvate kinase subtype M2. Int. J. Mol. Med. 2012, 30, 302–308. [Google Scholar] [CrossRef]
- Dey, P.; Kundu, A.; Sachan, R.; Park, J.H.; Ahn, M.Y.; Yoon, K.; Lee, J.; Kim, N.D.; Kim, I.S.; Lee, B.M.; et al. PKM2 Knockdown Induces Autophagic Cell Death via AKT/mTOR Pathway in Human Prostate Cancer Cells. Cell Physiol. Biochem. 2019, 52, 1535–1552. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, Z.; Li, G.; Lai, X.; Gu, R.; Xu, W.; Chen, H.; Xing, Z.; Chen, L.; Qian, J.; et al. Pyruvate Kinase M2 Promotes Prostate Cancer Metastasis Through Regulating ERK1/2-COX-2 Signaling. Front. Oncol. 2020, 10, 544288. [Google Scholar] [CrossRef]
- Lin, Y.; Lv, F.; Liu, F.; Guo, X.; Fan, Y.; Gu, F.; Gu, J.; Fu, L. High Expression of Pyruvate Kinase M2 is Associated with Chemosensitivity to Epirubicin and 5-Fluorouracil in Breast Cancer. J. Cancer 2015, 6, 1130–1139. [Google Scholar] [CrossRef]
- Guan, M.; Tong, Y.; Guan, M.; Liu, X.; Wang, M.; Niu, R.; Zhang, F.; Dong, D.; Shao, J.; Zhou, Y. Lapatinib Inhibits Breast Cancer Cell Proliferation by Influencing PKM2 Expression. Technol. Cancer Res. Treat. 2018, 17, 1533034617749418. [Google Scholar] [CrossRef]
- Ji, F.; Guo, B.; Wang, N.; Zhong, C.; Huang, L.; Huang, Y.; Wei, L.; Su, M.; Jiang, Y.; Jin, Q.; et al. Pyruvate kinase M2 interacts with mammalian sterile 20-like kinase 1 and inhibits tamoxifen-induced apoptosis in human breast cancer cells. Tumor Biol. 2017, 39, 1010428317692251. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Pulugu, P.; Singh, A.A.; Chatterjee, D.R.; Baviskar, S.; Vyas, H.; Behera, S.K.; Srivastava, A.; Kumar, H.; Shard, A. Mechanistic Investigation of Thiazole-Based Pyruvate Kinase M2 Inhibitor Causing Tumor Regression in Triple-Negative Breast Cancer. J. Med. Chem. 2024, 67, 3339–3357. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Oh, Y.; Lee, J.-S.; Park, J.H.; Shin, J.-K.; Han, J.-H.; Kim, H.S.; Yoon, S. Combination Treatment Using Pyruvate Kinase M2 Inhibitors for the Sensitization of High-Density Triple-negative Breast Cancer Cells. Vivo 2022, 36, 2105–2115. [Google Scholar] [CrossRef]
- Hasan, D.; Gamen, E.; Abu Tarboush, N.; Ismail, Y.; Pak, O.; Azab, B. PKM2 and HIF-1α regulation in prostate cancer cell lines. PLoS ONE 2018, 13, e0203745. [Google Scholar] [CrossRef]
- Jiang, C.; Zhao, X.; Jeong, T.; Kang, J.Y.; Park, J.H.; Kim, I.S.; Kim, H.S. Novel Specific Pyruvate Kinase M2 Inhibitor, Compound 3h, Induces Apoptosis and Autophagy through Suppressing Akt/mTOR Signaling Pathway in LNCaP Cells. Cancers 2022, 15, 265. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, L.; Chen, Y.; Zhou, C.; Wang, X.; Wang, D.; Liu, Z. PKM2 Promotes Breast Cancer Progression by Regulating Epithelial Mesenchymal Transition. Anal. Cell. Pathol. 2020, 2020, 8396023. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Zhang, X.; Yang, R.; Wei, X.; Yan, R.; Jiang, Y.; Shen, W. Expression Characteristics and Significant Prognostic Values of PGK1 in Breast Cancer. Front. Mol. Biosci. 2021, 8, 695420. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, J.D.; He, R.Q.; Huang, Z.G.; Chen, G.; Zou, W. Bibliometric analysis of phosphoglycerate kinase 1 expression in breast cancer and its distinct upregulation in triple-negative breast cancer. World J. Clin. Oncol. 2024, 15, 867–894. [Google Scholar] [CrossRef]
- Fu, D.; He, C.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. PGK1 is a Potential Survival Biomarker and Invasion Promoter by Regulating the HIF-1α–Mediated Epithelial-Mesenchymal Transition Process in Breast Cancer. Cell. Physiol. Biochem. 2018, 51, 2434–2444. [Google Scholar] [CrossRef]
- Xu, D.; Aka, J.A.; Wang, R.; Lin, S.X. 17beta-hydroxysteroid dehydrogenase type 5 is negatively correlated to apoptosis inhibitor GRP78 and tumor-secreted protein PGK1 and modulates breast cancer cell viability and proliferation. J. Steroid Biochem. Mol. Biol. 2017, 171, 270–280. [Google Scholar] [CrossRef]
- Sun, S.; Liang, X.; Zhang, X.; Liu, T.; Shi, Q.; Song, Y.; Jiang, Y.; Wu, H.; Jiang, Y.; Lu, X.; et al. Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer. Br. J. Cancer 2015, 112, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bai, Y.; Gao, Y.; Li, D.; Chen, L.; Zhou, C.; Feng, M.; Chen, X.; Jin, W.; Cao, Y. Systematic Analysis Uncovers Associations of PGK1 with Prognosis and Immunological Characteristics in Breast Cancer. Dis. Markers 2021, 2021, 7711151. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, R.; Wang, M.; Liu, T.; Zhou, Q.; Zhang, D.; Wang, J.; Shen, M.; Ren, X.; Sun, Q. PGAM1 regulation of ASS1 contributes to the progression of breast cancer through the cAMP/AMPK/CEBPB pathway. Mol. Oncol. 2022, 16, 2843–2860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, M.; Wang, W.; Ma, S.; Yu, W.; Ren, X.; Sun, Q. PGAM1 suppression remodels the tumor microenvironment in triple-negative breast cancer and synergizes with anti–PD-1 immunotherapy. J. Leukoc. Biol. 2024, 116, 579–588. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, M.; Ma, S.; Liu, M.; Yu, W.; Zhang, X.; Liu, T.; Liu, S.; Ren, X.; Sun, Q. Phosphoglycerate mutase 1 promotes breast cancer progression through inducing immunosuppressive M2 macrophages. Cancer Gene Ther. 2024, 31, 1018–1033. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, N.; Sun, W.; Li, X.; Liu, B.; Xie, Z.; Qu, J.; Xu, J.; Yang, X.; Su, Y.; et al. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene 2017, 36, 2900–2909. [Google Scholar] [CrossRef]
- Mele, L.; la Noce, M.; Paino, F.; Regad, T.; Wagner, S.; Liccardo, D.; Papaccio, G.; Lombardi, A.; Caraglia, M.; Tirino, V.; et al. Glucose-6-phosphate dehydrogenase blockade potentiates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. J. Exp. Clin. Cancer Res. 2019, 38, 160. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, F.; Zhang, Y.; Lin, Z.; Yang, J.; Han, X.; Feng, Y.; Pei, X.; Li, F.; Liu, Q.; et al. Targeting glucose-6-phosphate dehydrogenase by 6-AN induces ROS-mediated autophagic cell death in breast cancer. Febs. J. 2023, 290, 763–779. [Google Scholar] [CrossRef]
- Jhaveri, K.D.; Wanchoo, R.; Sakhiya, V.; Ross, D.W.; Fishbane, S. Adverse Renal Effects of Novel Molecular Oncologic Targeted Therapies: A Narrative Review. Kidney Int. Rep. 2017, 2, 108–123. [Google Scholar] [CrossRef]
- Zhen, X.; Zhang, M.; Hao, S.; Sun, J. Glucose-6-phosphate dehydrogenase and transketolase: Key factors in breast cancer progression and therapy. Biomed. Pharmacother. 2024, 176, 116935. [Google Scholar] [CrossRef]
- Révillion, F.; Pawlowski, V.; Hornez, L.; Peyrat, J.P. Glyceraldehyde-3-phosphate dehydrogenase gene expression in human breast cancer. Eur. J. Cancer 2000, 36, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Cho, W.C.S.; Ma, V.; Cheuk, W.; So, Y.K.; Wong, S.C.C.; Zhang, M.; Li, C.; Sun, Y.; Zhang, H.; et al. ADAM9 Mediates Triple-Negative Breast Cancer Progression via AKT/NF-κB Pathway. Front. Med. 2020, 7, 214. [Google Scholar] [CrossRef]
- Song, P.; Wu, J.; Chen, J.; Wang, F.; Chen, J.; Wang, G. Knockdown of circ-ADAM9 inhibits malignant phenotype and enhances radiosensitivity in breast cancer cells via acting as a sponge for miR-383-5p. Strahlenther. Und Onkol. 2023, 199, 78–89. [Google Scholar] [CrossRef]
- Cheng, Y.; Lin, L.; Li, X.; Lu, A.; Hou, C.; Wu, Q.; Hu, X.; Zhou, Z.; Chen, Z.; Tang, F. ADAM10 is involved in the oncogenic process and chemo-resistance of triple-negative breast cancer via regulating Notch1 signaling pathway, CD44 and PrPc. Cancer Cell Int. 2021, 21, 32. [Google Scholar] [CrossRef]
- Mullooly, M.; McGowan, P.M.; Kennedy, S.A.; Madden, S.F.; Crown, J.; O’ Donovan, N.; Duffy, M.J. ADAM10: A new player in breast cancer progression? Br. J. Cancer 2015, 113, 945–951. [Google Scholar] [CrossRef]
- Fry, J.L.; Toker, A. Secreted and Membrane-Bound Isoforms of Protease ADAM9 Have Opposing Effects on Breast Cancer Cell Migration. Cancer Res. 2010, 70, 8187–8198. [Google Scholar] [CrossRef]
- Micocci, K.C.; Martin, A.C.B.M.; Montenegro, C.d.F.; Durante, A.C.; Pouliot, N.; Cominetti, M.R.; Selistre-de-Araujo, H.S. ADAM9 silencing inhibits breast tumor cell invasion In Vitro. Biochimie 2013, 95, 1371–1378. [Google Scholar] [CrossRef]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.-L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a tumor biomarker and therapeutic target. Exp. Hematol. Oncol. 2020, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Scialò, C.; Legname, G. Chapter Twelve—The role of the cellular prion protein in the uptake and toxic signaling of pathological neurodegenerative aggregates. In Progress in Molecular Biology and Translational Science; Legname, G., Ed.; Academic Press: San Diego, CA, USA, 2020; Volume 175, pp. 297–323. [Google Scholar]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Lee, M.A.; Chan, T.-H.; Li, J.; Ni, Y.-B.; Shao, Y.; Chan, S.-K.; Cheungc, S.-Y.; Lau, K.-F.; Tse, G.M.K. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBiomedicine 2018, 38, 89–99. [Google Scholar] [CrossRef]
- Wang, Q.M.; Lv, L.; Tang, Y.; Zhang, L.; Wang, L.F. MMP-1 is overexpressed in triple-negative breast cancer tissues and the knockdown of MMP-1 expression inhibits tumor cell malignant behaviors In Vitro. Oncol. Lett. 2019, 17, 1732–1740. [Google Scholar] [CrossRef]
- Mohammadian, H.; Sharifi, R.; Rezanezhad Amirdehi, S.; Taheri, E.; Babazadeh Bedoustani, A. Matrix metalloproteinase MMP1 and MMP9 genes expression in breast cancer tissue. Gene Rep. 2020, 21, 100906. [Google Scholar] [CrossRef]
- Liu, H.; Kato, Y.; Erzinger, S.A.; Kiriakova, G.M.; Qian, Y.; Palmieri, D.; Steeg, P.S.; Price, J.E. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer 2012, 12, 583. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Q.; Hu, G.; Van Poznak, C.; Fleisher, M.; Reiss, M.; Massagué, J.; Kang, Y. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes. Dev. 2009, 23, 1882–1894. [Google Scholar] [CrossRef]
- Kim, H.W.; Park, J.E.; Baek, M.; Kim, H.; Ji, H.W.; Yun, S.H.; Jeong, D.; Ham, J.; Park, S.; Lu, X.; et al. Matrix Metalloproteinase-1 (MMP1) Upregulation through Promoter Hypomethylation Enhances Tamoxifen Resistance in Breast Cancer. Cancers 2022, 14, 1232. [Google Scholar] [CrossRef]
- Shen, C.-J.; Kuo, Y.-L.; Chen, C.-C.; Chen, M.-J.; Cheng, Y.-M. MMP1 expression is activated by Slug and enhances multi-drug resistance (MDR) in breast cancer. PLoS ONE 2017, 12, e0174487. [Google Scholar] [CrossRef]
- Bajor, M.; Zych, A.O.; Graczyk-Jarzynka, A.; Muchowicz, A.; Firczuk, M.; Trzeciak, L.; Gaj, P.; Domagala, A.; Siernicka, M.; Zagozdzon, A.; et al. Targeting peroxiredoxin 1 impairs growth of breast cancer cells and potently sensitises these cells to prooxidant agents. Br. J. Cancer 2018, 119, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Tehan, L.; Taparra, K.; Phelan, S. Peroxiredoxin overexpression in MCF-7 breast cancer cells and regulation by cell proliferation and oxidative stress. Cancer Investig. 2013, 31, 374–384. [Google Scholar] [CrossRef]
- McDonald, C.; Muhlbauer, J.; Perlmutter, G.; Taparra, K.; Phelan, S.A. Peroxiredoxin proteins protect MCF-7 breast cancer cells from doxorubicin-induced toxicity. Int. J. Oncol. 2014, 45, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-H.; Lian, X.-D.; Lee, S.-J.; Li, W.-L.; Sun, H.-N.; Jin, M.-H.; Kwon, T. Regulatory effect of peroxiredoxin 1 (PRDX1) on doxorubicin-induced apoptosis in triple negative breast cancer cells. Appl. Biol. Chem. 2022, 65, 63. [Google Scholar] [CrossRef]
- O’Leary, P.C.; Terrile, M.; Bajor, M.; Gaj, P.; Hennessy, B.T.; Mills, G.B.; Zagozdzon, A.; O’Connor, D.P.; Brennan, D.J.; Connor, K.; et al. Peroxiredoxin-1 protects estrogen receptor α from oxidative stress-induced suppression and is a protein biomarker of favorable prognosis in breast cancer. Breast Cancer Res. 2014, 16, R79. [Google Scholar] [CrossRef]
- Fiskus, W.; Coothankandaswamy, V.; Chen, J.; Ma, H.; Ha, K.; Saenz, D.T.; Krieger, S.S.; Mill, C.P.; Sun, B.; Huang, P.; et al. SIRT2 Deacetylates and Inhibits the Peroxidase Activity of Peroxiredoxin-1 to Sensitize Breast Cancer Cells to Oxidant Stress-Inducing Agents. Cancer Res. 2016, 76, 5467–5478. [Google Scholar] [CrossRef]
- Spínola-Lasso, E.; Montero, J.C.; Jiménez-Monzón, R.; Estévez, F.; Quintana, J.; Guerra, B.; Elokely, K.M.; León, F.; del Rosario, H.; Fernández-Pérez, L.; et al. Chemical-proteomics Identify Peroxiredoxin-1 as an Actionable Target in Triple-negative Breast Cancer. Int. J. Biol. Sci. 2023, 19, 1731–1747. [Google Scholar] [CrossRef]
- Stresing, V.; Baltziskueta, E.; Rubio, N.; Blanco, J.; Arriba, M.; Valls, J.; Janier, M.; Clézardin, P.; Sanz-Pamplona, R.; Nieva, C.; et al. Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene 2013, 32, 724–735. [Google Scholar] [CrossRef]
- Wang, T.; Tamae, D.; LeBon, T.; Shively, J.E.; Yen, Y.; Li, J.J. The Role of Peroxiredoxin II in Radiation-Resistant MCF-7 Breast Cancer Cells. Cancer Res. 2005, 65, 10338–10346. [Google Scholar] [CrossRef]
- Jin, X.; Wei, Y.; Xu, F.; Zhao, M.; Dai, K.; Shen, R.; Yang, S.; Zhang, N. SIRT1 promotes formation of breast cancer through modulating Akt activity. J. Cancer 2018, 9, 2012–2023. [Google Scholar] [CrossRef]
- Wang, D.; Li, C.; Zhang, X. The promoter methylation status and mRNA expression levels of CTCF and SIRT6 in sporadic breast cancer. DNA Cell Biol. 2014, 33, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Kumari, S.; Pulipaka, S.; Chandra, Y.; Kotamraju, S. SIRT1 promotes doxorubicin-induced breast cancer drug resistance and tumor angiogenesis via regulating GSH-mediated redox homeostasis. Mol. Carcinog. 2024, 63, 2291–2304. [Google Scholar] [CrossRef] [PubMed]
- Andreani, C.; Bartolacci, C.; Persico, G.; Casciaro, F.; Amatori, S.; Fanelli, M.; Giorgio, M.; Galié, M.; Tomassoni, D.; Wang, J.; et al. SIRT6 promotes metastasis and relapse in HER2-positive breast cancer. Sci. Rep. 2023, 13, 22000. [Google Scholar] [CrossRef]
- Nicknam, A.; Khojasteh Pour, S.; Hashemnejad, M.A.; Hussen, B.M.; Safarzadeh, A.; Eslami, S.; Taheri, M.; Ghafouri-Fard, S.; Jamali, E. Expression analysis of Rho GTPase-related lncRNAs in breast cancer. Pathol.-Res. Pract. 2023, 244, 154429. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Busch, H.; Boerries, M.; Brummer, T.; Timme, S.; Lassmann, S.; Aktories, K.; Schmidt, G. Specific role of RhoC in tumor invasion and metastasis. Oncotarget 2017, 8, 87364. [Google Scholar] [CrossRef]
- Kleer, C.G.; van Golen, K.L.; Zhang, Y.; Wu, Z.-F.; Rubin, M.A.; Merajver, S.D. Characterization of RhoC Expression in Benign and Malignant Breast Disease: A Potential New Marker for Small Breast Carcinomas with Metastatic Ability. Am. J. Pathol. 2002, 160, 579–584. [Google Scholar] [CrossRef]
- Pillé, J.Y.; Denoyelle, C.; Varet, J.; Bertrand, J.R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.L.; Vannier, J.P.; Malvy, C.; et al. Anti-RhoA and Anti-RhoC siRNAs Inhibit the Proliferation and Invasiveness of MDA-MB-231 Breast Cancer Cells In Vitro and In Vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Z.F.; Rosenthal, D.T.; Rhee, E.M.; Merajver, S.D. Characterization of the roles of RHOC and RHOA GTPases in invasion, motility, and matrix adhesion in inflammatory and aggressive breast cancers. Cancer 2010, 116, 2768–2782. [Google Scholar] [CrossRef]
- Xu, X.D.; Shen, H.B.; Zhu, L.; Lu, J.Q.; Zhang, L.; Luo, Z.Y.; Wu, Y.Q. Anti-RhoC siRNAs inhibit the proliferation and invasiveness of breast cancer cells via modulating the KAI1, MMP9, and CXCR4 expression. Onco Targets Ther. 2017, 10, 1827–1834. [Google Scholar] [CrossRef]
- Kalpana, G.; Figy, C.; Yeung, M.; Yeung, K.C. Reduced RhoA expression enhances breast cancer metastasis with a concomitant increase in CCR5 and CXCR4 chemokines signaling. Sci. Rep. 2019, 9, 16351. [Google Scholar] [CrossRef]
- Asghar, K.; Loya, A.; Rana, I.A.; Tahseen, M.; Ishaq, M.; Farooq, A.; Bakar, M.A.; Masood, I. Indoleamine 2,3-dioxygenase expression and overall survival in patients diagnosed with breast cancer in Pakistan. Cancer Manag. Res. 2019, 11, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Bilir, C.; Eskiler, G.G.; Bilir, F. The cytotoxic effects of indoleamine 2, 3-dioxygenase inhibitors on triple negative breast cancer cells upon tumor necrosis factor α stimulation. J. Cancer Res. Ther. 2023, 19, S74–S80. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhu, S.; Li, M.; Li, F.; Wei, F.; Liu, J.; Ren, X. High Indoleamine 2,3-Dioxygenase Is Correlated With Microvessel Density and Worse Prognosis in Breast Cancer. Front. Immunol. 2018, 9, 724. [Google Scholar] [CrossRef]
- Zhao, X.; Jiang, Y.; Xu, M.; Hu, J.; Feng, N.; Deng, H.; Lu, C.; Huang, T. Indoleamine 2,3-dioxygenase 1 regulates breast cancer tamoxifen resistance through interleukin-6/signal transducer and activator of transcription. Toxicol. Appl. Pharmacol. 2022, 440, 115921. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, L.; Liu, J.; Li, F. Chemoresistance was correlated with elevated expression and activity of indoleamine 2,3-dioxygenase in breast cancer. Cancer Chemother. Pharmacol. 2020, 85, 77–93. [Google Scholar] [CrossRef]
- Sarangi, P. Role of indoleamine 2, 3-dioxygenase 1 in immunosuppression of breast cancer. Cancer Pathog. Ther. 2024, 2, 246–255. [Google Scholar] [CrossRef]
- Yu, J.; Sun, J.; Wang, S.E.; Li, H.; Cao, S.; Cong, Y.; Liu, J.; Ren, X. Upregulated Expression of Indoleamine 2, 3-Dioxygenase in Primary Breast Cancer Correlates with Increase of Infiltrated Regulatory T Cells In Situ and Lymph Node Metastasis. J. Immunol. Res. 2011, 2011, 469135. [Google Scholar] [CrossRef]
- Levina, V.; Su, Y.; Gorelik, E. Immunological and Nonimmunological Effects of Indoleamine 2,3-Dioxygenase on Breast Tumor Growth and Spontaneous Metastasis Formation. J. Immunol. Res. 2012, 2012, 173029. [Google Scholar] [CrossRef]
- Xu, D.; Xu, N.; Sun, L.; Yang, Z.; He, M.; Li, Y. TG2 as a novel breast cancer prognostic marker promotes cell proliferation and glycolysis by activating the MEK/ERK/LDH pathway. BMC Cancer 2022, 22, 1267. [Google Scholar] [CrossRef]
- Hettasch, J.M.; Bandarenko, N.; Burchette, J.L.; Lai, T.S.; Marks, J.R.; Haroon, Z.A.; Peters, K.; Dewhirst, M.W.; Iglehart, J.D.; Greenberg, C.S. Tissue transglutaminase expression in human breast cancer. Lab. Investig. 1996, 75, 637–645. [Google Scholar] [PubMed]
- Mehta, K.; Fok, J.; Miller, F.R.; Koul, D.; Sahin, A.A. Prognostic Significance of Tissue Transglutaminase in Drug Resistant and Metastatic Breast Cancer. Clin. Cancer Res. 2004, 10, 8068–8076. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.F.; Mangala, L.S.; Mehta, K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene 2006, 25, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Wang, X.H.; Hua, Y.T.; Zhang, Y.Z.; Han, Y.; Yang, Z.L. The tissue transglutaminase: A potential target regulating MDR in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6175–6184. [Google Scholar] [CrossRef]
- Gallo, M.; Ferrari, E.; Terrazzan, A.; Brugnoli, F.; Spisni, A.; Taccioli, C.; Aguiari, G.; Trentini, A.; Volinia, S.; Keillor, J.W.; et al. Metabolic characterisation of transglutaminase 2 inhibitor effects in breast cancer cell lines. FEBS J. 2023, 290, 5411–5433. [Google Scholar] [CrossRef]
- Mangala, L.S.; Fok, J.Y.; Zorrilla-Calancha, I.R.; Verma, A.; Mehta, K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 2007, 26, 2459–2470. [Google Scholar] [CrossRef]
- Seo, S.; Moon, Y.; Choi, J.; Yoon, S.; Jung, K.H.; Cheon, J.; Kim, W.; Kim, D.; Lee, C.H.; Kim, S.W.; et al. The GTP binding activity of transglutaminase 2 promotes bone metastasis of breast cancer cells by downregulating microRNA-205. Am. J. Cancer Res. 2019, 9, 597–607. [Google Scholar]
- Schwager, S.C.; Young, K.M.; Hapach, L.A.; Carlson, C.M.; Mosier, J.A.; McArdle, T.J.; Wang, W.; Schunk, C.; Jayathilake, A.L.; Bates, M.E.; et al. Weakly migratory metastatic breast cancer cells activate fibroblasts via microvesicle-Tg2 to facilitate dissemination and metastasis. Elife 2022, 11, e74433. [Google Scholar] [CrossRef]
- Tatarov, O.; Mitchell, T.J.; Seywright, M.; Leung, H.Y.; Brunton, V.G.; Edwards, J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin. Cancer Res. 2009, 15, 3540–3549. [Google Scholar] [CrossRef]
- Xu, W.; Allbritton, N.; Lawrence, D.S. Src Kinase Regulation in Progressively Invasive Cancer. PLoS ONE 2012, 7, e48867. [Google Scholar] [CrossRef]
- Gelman, I.H.; Peresie, J.; Eng, K.H.; Foster, B.A. Differential Requirement for Src Family Tyrosine Kinases in the Initiation, Progression, and Metastasis of Prostate Cancer. Mol. Cancer Res. 2014, 12, 1470–1479. [Google Scholar] [CrossRef]
- Cai, H.; Babic, I.; Wei, X.; Huang, J.; Witte, O.N. Invasive Prostate Carcinoma Driven by c-Src and Androgen Receptor Synergy. Cancer Res. 2011, 71, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, J.S.; Bond, D.R.; Jankowski, H.; Goldie, B.J.; Burchell, R.; Naudin, C.; Smith, N.D.; Scarlett, C.J.; Larsen, M.R.; Dun, M.D.; et al. Extracellular vesicles with altered tetraspanin CD9 and CD151 levels confer increased prostate cell motility and invasion. Sci. Rep. 2018, 8, 8822. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Siddiqui, I.A.; Hafeez, B.B.; Baniahmad, A.; Mukhtar, H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene 2008, 27, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, I.; Wang, J.; Qin, M.; Gao, L.; Holtz, R.; Vessella, R.L.; Leach, R.W.; Gelman, I.H. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 2016, 8, 10324. [Google Scholar]
- Goc, A.; Al-Husein, B.A.-H.; Katsanevas, K.; Steinbach, A.; Lou, U.; Sabbineni, H.; DeRemer, D.L.; Somanath, P.R. Targeting Src-mediated Tyr216 phosphorylation and activation of GSK-3 in prostate cancer cells inhibit prostate cancer progression In Vitro and In Vivo. Oncotarget 2014, 5, 775. [Google Scholar]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Chang, Y.M.; Bai, L.; Liu, S.; Yang, J.C.; Kung, H.J.; Evans, C.P. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene 2008, 27, 6365–6375. [Google Scholar] [CrossRef]
- Yang, C.-C.; Fazli, L.; Loguercio, S.; Zharkikh, I.; Aza-Blanc, P.E.; Gleave, M.; Wolf, D.A. Downregulation of c-SRC kinase CSK promotes castration resistant prostate cancer and pinpoints a novel disease subclass. Oncotarget 2015, 6, 22060. [Google Scholar]
- Wu, Z.; Chang, P.-C.; Yang, J.C.; Chu, C.-Y.; Wang, L.-Y.; Chen, N.-T.; Ma, A.-H.; Desai, S.J.; Lo, S.H.; Evans, C.P.; et al. Autophagy Blockade Sensitizes Prostate Cancer Cells towards Src Family Kinase Inhibitors. Genes. Cancer 2010, 1, 40–49. [Google Scholar] [CrossRef]
- Le Page, C.; Koumakpayi, I.H.; Alam-Fahmy, M.; Mes-Masson, A.M.; Saad, F. Expression and localisation of Akt-1, Akt-2 and Akt-3 correlate with clinical outcome of prostate cancer patients. Br. J. Cancer 2006, 94, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; McNulty, A.M.; Wang, Z.; Houck, K.; Allen, S.; Paul, J.D.; Hbaiu, A.; Goode, R.G.; Sandusky, G.E.; et al. Increased AKT Activity Contributes to Prostate Cancer Progression by Dramatically Accelerating Prostate Tumor Growth and Diminishing p27Kip1 Expression. J. Biol. Chem. 2000, 275, 24500–24505. [Google Scholar] [CrossRef] [PubMed]
- Goc, A.; Liu, J.; Byzova, T.V.; Somanath, P.R. Akt1 mediates prostate cancer cell microinvasion and chemotaxis to metastatic stimuli via integrin β3 affinity modulation. Br. J. Cancer 2012, 107, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Dong, B.; He, X.; Qiu, Z.; Zhang, J.; Zhang, M.; Liu, H.; Pang, X.; Cui, Y. The challenges and opportunities of αvβ3-based therapeutics in cancer: From bench to clinical trials. Pharmacol. Res. 2023, 189, 106694. [Google Scholar] [CrossRef]
- Alasmar, A.; Al-Alami, Z.; Zein, S.; Al-Smadi, A.; Al Bashir, S.; Alorjani, M.S.; Al-Zoubi, R.M.; Al Zoubi, M. Novel Mutations in AKT1 Gene in Prostate Cancer Patients in Jordan. Curr. Issues Mol. Biol. 2024, 46, 9856–9866. [Google Scholar] [CrossRef]
- Vaarala, M.H.; Porvari, K.; Kyllönen, A.; Vihko, P. Differentially expressed genes in two LNCaP prostate cancer cell lines reflecting changes during prostate cancer progression. Lab. Investig. 2000, 80, 1259–1268. [Google Scholar] [CrossRef]
- Vaarala, M.H.; Porvari, K.; Kyllönen, A.; Lukkarinen, O.; Vihko, P. The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: Detection of mutated TMPRSS2 form in a case of aggressive disease. Int. J. Cancer 2001, 94, 705–710. [Google Scholar] [CrossRef]
- Chen, Y.W.; Lee, M.S.; Lucht, A.; Chou, F.P.; Huang, W.; Havighurst, T.C.; Kim, K.; Wang, J.K.; Antalis, T.M.; Johnson, M.D.; et al. TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am. J. Pathol. 2010, 176, 2986–2996. [Google Scholar] [CrossRef]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef]
- Ko, C.-J.; Huang, C.-C.; Lin, H.-Y.; Juan, C.-P.; Lan, S.-W.; Shyu, H.-Y.; Wu, S.-R.; Hsiao, P.-W.; Huang, H.-P.; Shun, C.-T.; et al. Androgen-Induced TMPRSS2 Activates Matriptase and Promotes Extracellular Matrix Degradation, Prostate Cancer Cell Invasion, Tumor Growth, and Metastasis. Cancer Res. 2015, 75, 2949–2960. [Google Scholar] [CrossRef]
- Tsai, C.H.; Teng, C.H.; Tu, Y.T.; Cheng, T.S.; Wu, S.R.; Ko, C.J.; Shyu, H.Y.; Lan, S.W.; Huang, H.P.; Tzeng, S.F.; et al. HAI-2 suppresses the invasive growth and metastasis of prostate cancer through regulation of matriptase. Oncogene 2014, 33, 4643–4652. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R. TMPRSS2 promotes metastasis through proteolysis. Nat. Rev. Urol. 2014, 11, 546. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.-J.; Hsu, T.-W.; Wu, S.-R.; Lan, S.-W.; Hsiao, T.-F.; Lin, H.-Y.; Lin, H.-H.; Tu, H.-F.; Lee, C.-F.; Huang, C.-C.; et al. Inhibition of TMPRSS2 by HAI-2 reduces prostate cancer cell invasion and metastasis. Oncogene 2020, 39, 5950–5963. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bujanda, Z.A.; Obradovic, A.; Nirschl, T.R.; Crowley, L.; Macedo, R.; Papachristodoulou, A.; O’Donnell, T.; Laserson, U.; Zarif, J.C.; Reshef, R.; et al. TGM4: An immunogenic prostate-restricted antigen. J. Immunother. Cancer 2021, 9, e001649. [Google Scholar] [CrossRef]
- Davies, G.; Ablin, R.J.; Mason, M.D.; Jiang, W.G. Expression of the prostate transglutaminase (TGase-4) in prostate cancer cells and its impact on the invasiveness of prostate cancer. J. Exp. Ther. Oncol. 2007, 6, 257–264. [Google Scholar]
- Jiang, W.G.; Ye, L.; Ablin, R.J.; Kynaston, H.G.; Mason, M.D. The prostate transglutaminase, TGase-4, coordinates with the HGFL/MSP-RON system in stimulating the migration of prostate cancer cells. Int. J. Oncol. 2010, 37, 413–418. [Google Scholar] [CrossRef]
- Jiang, W.G.; Ye, L.; Sanders, A.J.; Ruge, F.; Kynaston, H.G.; Ablin, R.J.; Mason, M.D. Prostate transglutaminase (TGase-4, TGaseP) enhances the adhesion of prostate cancer cells to extracellular matrix, the potential role of TGase-core domain. J. Transl. Med. 2013, 11, 269. [Google Scholar] [CrossRef]
- Ablin, R.J.; Kynaston, H.G.; Mason, M.D.; Jiang, W.G. Prostate transglutaminase (TGase-4) antagonizes the anti-tumour action of MDA-7/IL-24 in prostate cancer. J. Transl. Med. 2011, 9, 49. [Google Scholar] [CrossRef]
- Menezes, M.E.; Bhatia, S.; Bhoopathi, P.; Das, S.K.; Emdad, L.; Dasgupta, S.; Dent, P.; Wang, X.Y.; Sarkar, D.; Fisher, P.B. MDA-7/IL-24: Multifunctional cancer killing cytokine. Adv. Exp. Med. Biol. 2014, 818, 127–153. [Google Scholar] [CrossRef]
- Ablin, R.J.; Owen, S.; Jiang, W.G. Prostate Transglutaminase (TGase-4) Induces Epithelial-to-Mesenchymal Transition in Prostate Cancer Cells. Anticancer. Res. 2017, 37, 481–487. [Google Scholar] [CrossRef]
- Gomes, I.M.; Santos, C.R.; Maia, C.J. Expression of STEAP1 and STEAP1B in prostate cell lines, and the putative regulation of STEAP1 by post-transcriptional and post-translational mechanisms. Genes. Cancer 2014, 5, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Challita-Eid, P.M.; Morrison, K.; Etessami, S.; An, Z.; Morrison, K.J.; Perez-Villar, J.J.; Raitano, A.B.; Jia, X.-C.; Gudas, J.M.; Kanner, S.B.; et al. Monoclonal Antibodies to Six-Transmembrane Epithelial Antigen of the Prostate-1 Inhibit Intercellular Communication In Vitro and Growth of Human Tumor Xenografts In Vivo. Cancer Res. 2007, 67, 5798–5805. [Google Scholar] [CrossRef] [PubMed]
- Burnell, S.E.A.; Spencer-Harty, S.; Howarth, S.; Bodger, O.; Kynaston, H.; Morgan, C.; Doak, S.H. Utilisation of the STEAP protein family in a diagnostic setting may provide a more comprehensive prognosis of prostate cancer. PLoS ONE 2019, 14, e0220456. [Google Scholar] [CrossRef]
- Rocha, S.M.; Nascimento, D.; Coelho, R.S.; Cardoso, A.M.; Passarinha, L.A.; Socorro, S.; Maia, C.J. STEAP1 Knockdown Decreases the Sensitivity of Prostate Cancer Cells to Paclitaxel, Docetaxel and Cabazitaxel. Int. J. Mol. Sci. 2023, 24, 6643. [Google Scholar] [CrossRef]
- Jones, L.A.; Conway, G.E.; Nguyen-Chi, A.; Burnell, S.; Jenkins, G.J.; Conlan, R.S.; Doak, S.H. Investigating STEAP2 as a potential therapeutic target for the treatment of aggressive prostate cancer. Cell. Mol. Biol. 2023, 69, 179–187. [Google Scholar] [CrossRef]
- Burnell, S.E.A.; Spencer-Harty, S.; Howarth, S.; Bodger, O.; Kynaston, H.; Morgan, C.; Doak, S.H. STEAP2 Knockdown Reduces the Invasive Potential of Prostate Cancer Cells. Sci. Rep. 2018, 8, 6252. [Google Scholar] [CrossRef]
- Patel, S.; Turner, P.R.; Stubberfield, C.; Barry, E.; Rohlff, C.R.; Stamps, A.; Tyson, K.; Terrett, J.; Box, G.; Eccles, S.; et al. Hyaluronidase gene profiling and role of HYAL-1 overexpression in an orthotopic model of prostate cancer. Int. J. Cancer 2002, 97, 416–424. [Google Scholar] [CrossRef]
- Kovar, J.L.; Johnson, M.A.; Volcheck, W.M.; Chen, J.; Simpson, M.A. Hyaluronidase Expression Induces Prostate Tumor Metastasis in an Orthotopic Mouse Model. Am. J. Pathol. 2006, 169, 1415–1426. [Google Scholar] [CrossRef]
- Reale, A.; Khong, T.; Spencer, A. Extracellular Vesicles and Their Roles in the Tumor Immune Microenvironment. J. Clin. Med. 2022, 11, 6892. [Google Scholar] [CrossRef]
- Hamed, M.A.; Wasinger, V.; Wang, Q.; Graham, P.; Malouf, D.; Bucci, J.; Li, Y. Prostate cancer-derived extracellular vesicles metabolic biomarkers: Emerging roles for diagnosis and prognosis. J. Control. Release 2024, 371, 126–145. [Google Scholar] [CrossRef]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.-B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, H.; Cui, Y.; Xing, J.; Wang, W.; Chen, J.; Wang, S.; Yang, Z. Extracellular vesicles for breast cancer diagnosis and therapy. Extracell. Vesicle 2024, 3, 100039. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Sheykhhasan, M.; Kalhor, N.; Sheikholeslami, A.; Dolati, M.; Amini, E.; Fazaeli, H. Exosomes of Mesenchymal Stem Cells as a Proper Vehicle for Transfecting miR-145 into the Breast Cancer Cell Line and Its Effect on Metastasis. BioMed Res. Int. 2021, 2021, 5516078. [Google Scholar] [CrossRef]
- Huang, L.; Rong, Y.; Tang, X.; Yi, K.; Qi, P.; Hou, J.; Liu, W.; He, Y.; Gao, X.; Yuan, C.; et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer 2022, 21, 45. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, A.; de Miguel-Pérez, D.; Ortega, F.G.; García-Puche, J.L.; Robles-Fernández, I.; Exposito, J.; Martorell-Marugan, J.; Carmona-Sáez, P.; Garrido-Navas, M.D.C.; Rolfo, C.; et al. Exosomal miRNA profile as complementary tool in the diagnostic and prediction of treatment response in localized breast cancer under neoadjuvant chemotherapy. Breast Cancer Res. 2019, 21, 21. [Google Scholar] [CrossRef]
- Lim, J.; Kang, B.; Son, H.Y.; Mun, B.; Huh, Y.-M.; Rho, H.W.; Kang, T.; Moon, J.; Lee, J.-J.; Seo, S.B.; et al. Microfluidic device for one-step detection of breast cancer-derived exosomal mRNA in blood using signal-amplifiable 3D nanostructure. Biosens. Bioelectron. 2022, 197, 113753. [Google Scholar] [CrossRef]
- Bamankar, S.; Londhe, V.Y. The Rise of Extracellular Vesicles as New Age Biomarkers in Cancer Diagnosis: Promises and Pitfalls. Technol. Cancer Res. Treat. 2023, 22, 15330338221149266. [Google Scholar] [CrossRef]
- Rodríguez, M.; Bajo-Santos, C.; Hessvik, N.P.; Lorenz, S.; Fromm, B.; Berge, V.; Sandvig, K.; Linē, A.; Llorente, A. Identification of non-invasive miRNAs biomarkers for prostate cancer by deep sequencing analysis of urinary exosomes. Mol. Cancer 2017, 16, 156. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef]
- Andre, M.; Caobi, A.; Miles, J.S.; Vashist, A.; Ruiz, M.A.; Raymond, A.D. Diagnostic potential of exosomal extracellular vesicles in oncology. BMC Cancer 2024, 24, 322. [Google Scholar] [CrossRef]
- Dudzik, D.; Macioszek, S.; Struck-Lewicka, W.; Kordalewska, M.; Buszewska-Forajta, M.; Waszczuk-Jankowska, M.; Wawrzyniak, R.; Artymowicz, M.; Raczak-Gutknecht, J.; Siluk, D.; et al. Perspectives and challenges in extracellular vesicles untargeted metabolomics analysis. TrAC Trends Anal. Chem. 2021, 143, 116382. [Google Scholar] [CrossRef]
- Davidson, S.M.; Boulanger, C.M.; Aikawa, E.; Badimon, L.; Barile, L.; Binder, C.J.; Brisson, A.; Buzas, E.; Emanueli, C.; Jansen, F.; et al. Methods for the identification and characterization of extracellular vesicles in cardiovascular studies: From exosomes to microvesicles. Cardiovasc. Res. 2022, 119, 45–63. [Google Scholar] [CrossRef]
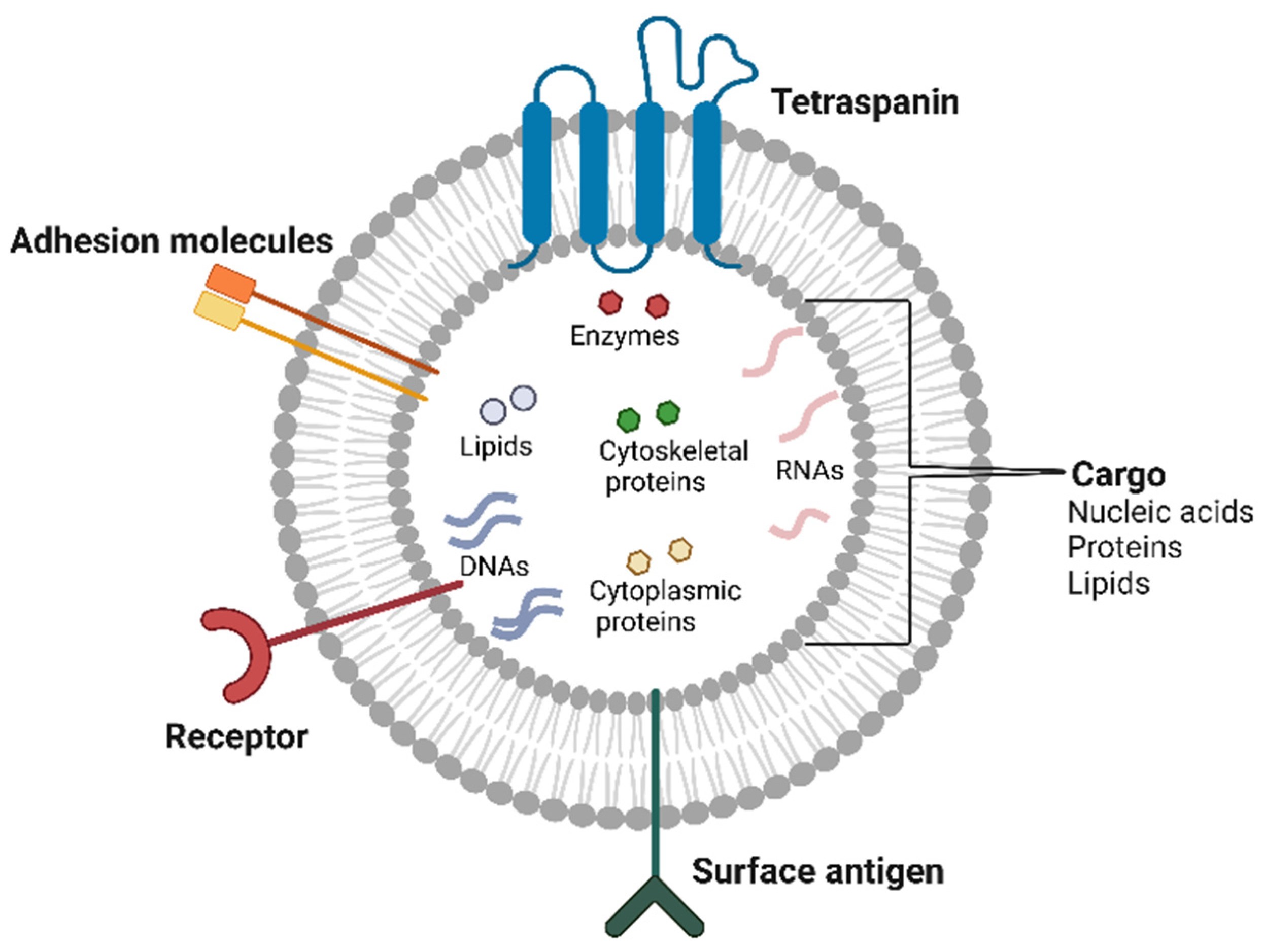
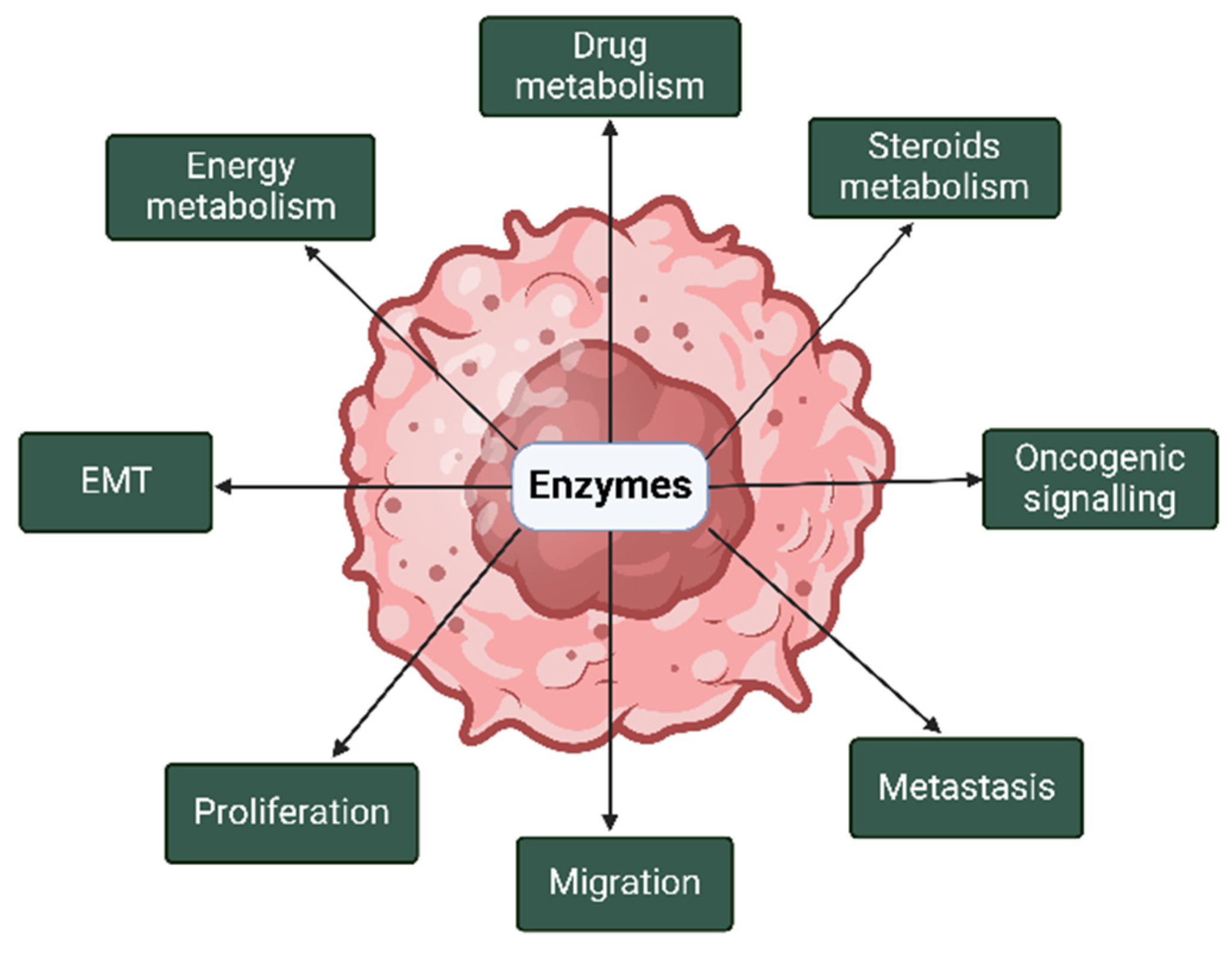



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Detection/Quantitation Method | Biological Function | Source of EVs | Malignant Functions | References |
|---|---|---|---|---|---|
| ATP citrate lyase | Protein-based (LC-MS/MS analysis, Western blotting, and ACLY activity assay) | Converts citrate to acetyl-CoA and oxaloacetate in the presence of coenzyme A and ATP | MCF-7 and MDA-MB-231 breast cancer cells | Promotes survival of breast cancer | [84] |
| Enolase 1 and 2 | Protein-based (LC-MS/MS analysis and Western blotting) | Converts 2-phosphoglycerate (2-PG) to phosphoenolpyruvate (PEP) | MCF-7, MDA-MB-231, and T-47D breast cancer cells | Promotes progression and survival of breast cancer | [85,86,87] |
| Fatty acid synthase | Protein-based (LC-MS/MS analysis and Western blotting) | Synthesizes the fatty acid palmitate from acetyl-CoA and malonyl-CoA in the presence of NADPH | MCF-7 breast cancer cells | Promotes progression and survival of breast cancer | [87] |
| Focal adhesion kinase | Protein-based (Western blotting) | Phosphorylates the OH group of tyrosine amino acid residue of specific protein targets | Peripheral blood from patients with breast cancer | Promotes progression and survival of breast cancer | [88] |
| Pyruvate kinase M2 | Protein-based (LC-MS/MS analysis and Western blotting) and mRNA-based (Double-tagging RT-PCR) | Transfers a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), producing pyruvate and ATP | MCF-7 and MDA-MB-231 breast cancer cells | Promotes progression and survival of breast cancer | [86,87] |
| Phosphoglycerate kinase 1 | mRNA-based (Double-tagging RT-PCR) | Reversibly transfers a phosphate group from 1,3-bisphosphoglycerate (1,3-BPG) to ADP, producing 3-phosphoglycerate (3-PG) and ATP | MCF-7 and MDA-MB-231 breast cancer cells | Promotes progression and survival of breast cancer | [86] |
| Phosphoglycerate mutase 1 | mRNA-based (Double-tagging RT-PCR) | Converts 3-phosphoglycerate (3PG) to 2-phosphoglycerate (2PG) through a 2,3-bisphosphoglycerate intermediate | MCF-7 and MDA-MB-231 breast cancer cells | Promotes progression and survival of breast cancer | [86] |
| Glucose-6-phosphate dehydrogenase | Protein-based (LC-MS/MS analysis and Western blotting) | Converts glucose 6-phosphate in the presence of nicotinamide adenine dinucleotide phosphate (oxidized) to 6-phospho-D-glucono-1,5-lactone, producing NADPH | MCF-7 breast cancer cells | Promotes cell survival of breast cancer | [87] |
| Glyceraldehyde 3-phosphate dehydrogenase | Protein-based (LC-MS/MS analysis and Western blotting) and mRNA-based (Double-tagging RT-PCR) | Converts glyceraldehyde 3-phosphate to D-glycerate 1,3-bisphosphate | MCF-7 and MDA-MB-231 breast cancer cells; plasma from patients with breast cancer | Promotes survival of breast cancer | [86,87,89] |
| A Disintegrin and Metalloproteinase 9 and 10 | Protein-based (LC-MS/MS analysis and Western blotting) | Cleaves extracellular portions of transmembrane proteins to release soluble ectodomains from the cell surface | Hs578T, BT-549, MDA-MB-231, LM2 breast cancer cells; plasma of patients with breast cancer | Promotes progression and survival of breast cancer | [90,91,92] |
| Matrix metalloproteinase 1 | Protein-based (LC-MS/MS analysis and Western blotting) | Degrades extracellular matrix proteins, such as gelatin and collagen IV and V. | MDA-MB-231-HM breast cancer cells; urine of patients with breast cancer | Promotes progression of breast cancer | [93,94] |
| Peroxiredoxin 1 and 2 | Protein-based (LC-MS/MS analysis and Western blotting) | Reduces hydrogen peroxides and different alkyl hydroperoxides to water | MDA-MB-231 breast cancer cells | Promotes progression and survival of breast cancer | [95] |
| Sirtuin 1 and 6 | Protein-based (LC-MS/MS analysis, Western blotting, and SIRT1 and SIRT6 activity assay) | Deacetylates protein targets and/or adds ADP-ribose moieties to protein targets | MCF-7 and MDA-MB-231 breast cancer cells | Promotes progression of breast cancer | [84] |
| RhoA/RhoC GTPase | Protein-based (Western blotting) | Binds and hydrolyses nucleotide guanosine triphosphate to guanosine diphosphate | MDA-MB-231, SK-BR-3, and HCC70 breast cancer cells | Promotes progression and survival of breast cancer | [96] |
| Indoleamine-2,3-dioxygenase | Protein-based (Western blotting) and mRNA-based (RT-PCR) | Oxidizes L-tryptophan to form N-formylkynurenine | Plasma from patients with breast cancer | Promotes progression and survival of breast cancer | [97] |
| Tissue transglutaminase | Protein-based (Western blotting and transamidation activity) | Crosslinks proteins between an ε-amino group of lysine and a γ-carboxamide group of glutamine, forming a proteolytically resistant inter- or intramolecular bond | MDA-MB-231 breast cancer cells | Promotes survival of breast cancer | [98] |
| Enzyme | Detection/Quantitation Method | Biological Function | Source of EVs | Malignant Functions | References |
|---|---|---|---|---|---|
| ATP citrate lyase | Protein-based (Western blotting and LC-QTOF MS analysis) | Converts citrate to acetyl-CoA and oxaloacetate in the presence of coenzyme A and ATP | DU145, VCaP, and LNCaP prostate cancer cells | Promotes survival of prostate cancer | [99] |
| Enolase 1 and 2 | Protein-based (Western blotting and LC-QTOF MS analysis) | Converts 2-phosphoglycerate (2-PG) to phosphoenolpyruvate (PEP) | DU145 and VCaP prostate cancer cells | Promotes progression and survival of prostate cancer | [99] |
| Fatty acid synthase | Protein-based (Western blotting and LC-QTOF MS analysis) | Converts acetyl-CoA and malonyl-CoA to a 16-carbon fatty acid palmitate | DU145, VCaP, LNCaP, and C4–2 prostate cancer cells | Promotes progression and survival of prostate cancer | [99] |
| Focal adhesion kinase | Protein-based (Western blotting) | Phosphorylates the OH group of tyrosine amino acid residue of protein targets | PC-3 prostate cancer cells | Promotes progression and survival of prostate cancer | [100] |
| Pyruvate kinase M2 | Protein-based (Western blotting and LC-QTOF MS analysis) | Converts phosphoenolpyruvate and ADP into pyruvate and ATP | VCaP, C4–2, LNCaP, DU145, and PC-3 prostate cancer cells | Promotes progression and survival of prostate cancer | [99,101] |
| Src kinase | Protein-based (Western blotting) | Phosphorylates the OH group of tyrosine amino acid residue of specific protein targets | C4-2B, PC-3, and DU145 prostate cancer cells | Promotes progression and survival of prostate cancer | [100] |
| Akt1 kinase | Protein-based (Western blotting and Akt kinase activity assay) | Phosphorylates the OH group of serine or threonine amino acid residue of protein targets | Plasma of patients with metastatic prostate cancer | Promotes progression of prostate cancer | [102] |
| TMPRSS2 serine protease | Protein-based (Western blotting and Akt Kinase activity assay) and mRNA-based (RT-PCR) | Cleaves peptide bonds in protein targets | PC-3 and LNCaP prostate cancer cells; and Urine/Plasma of patients with prostate cancer | Promotes progression of prostate cancer | [103,104,105] |
| Transglutaminase-4 | Protein-based (Western blotting) and mRNA-based (RT-PCR) | Forms an isopeptide bond between γ-carboxamide groups (-(C=O)NH2) of glutamine residue side chains and the ε-amino groups (-NH2) of lysine residue side chains | Urine of patients with prostate cancer | Promotes progression of prostate cancer | [104,105,106] |
| STEAP1 and 2 | Protein-based (Western blotting) and mRNA-based (RT-PCR) | Catalyzes the reduction of Iron and Copper | Plasma and Urine of patients with prostate cancer | Promotes progression and survival of prostate cancer | [104,107] |
| Hyaluronidase 1 | Protein-based (Western blotting) | Cleaves β1→4-N-acetylglucosaminide bonds of intracellular hyaluronan of all sizes | 22Rv1 human prostate adenocarcinoma cells | Promotes progression of prostate cancer | [108,109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onyiba, C.I.; Kumar, N.K.; Scarlett, C.J.; Weidenhofer, J. Cell Progression and Survival Functions of Enzymes Secreted in Extracellular Vesicles Associated with Breast and Prostate Cancers. Cells 2025, 14, 468. https://doi.org/10.3390/cells14070468
Onyiba CI, Kumar NK, Scarlett CJ, Weidenhofer J. Cell Progression and Survival Functions of Enzymes Secreted in Extracellular Vesicles Associated with Breast and Prostate Cancers. Cells. 2025; 14(7):468. https://doi.org/10.3390/cells14070468
Chicago/Turabian StyleOnyiba, Cosmos Ifeanyi, Niwasini Krishna Kumar, Christopher J. Scarlett, and Judith Weidenhofer. 2025. "Cell Progression and Survival Functions of Enzymes Secreted in Extracellular Vesicles Associated with Breast and Prostate Cancers" Cells 14, no. 7: 468. https://doi.org/10.3390/cells14070468
APA StyleOnyiba, C. I., Kumar, N. K., Scarlett, C. J., & Weidenhofer, J. (2025). Cell Progression and Survival Functions of Enzymes Secreted in Extracellular Vesicles Associated with Breast and Prostate Cancers. Cells, 14(7), 468. https://doi.org/10.3390/cells14070468