
Ciliary Ion Channels in Polycystic Kidney Disease


Abstract
1. Introduction
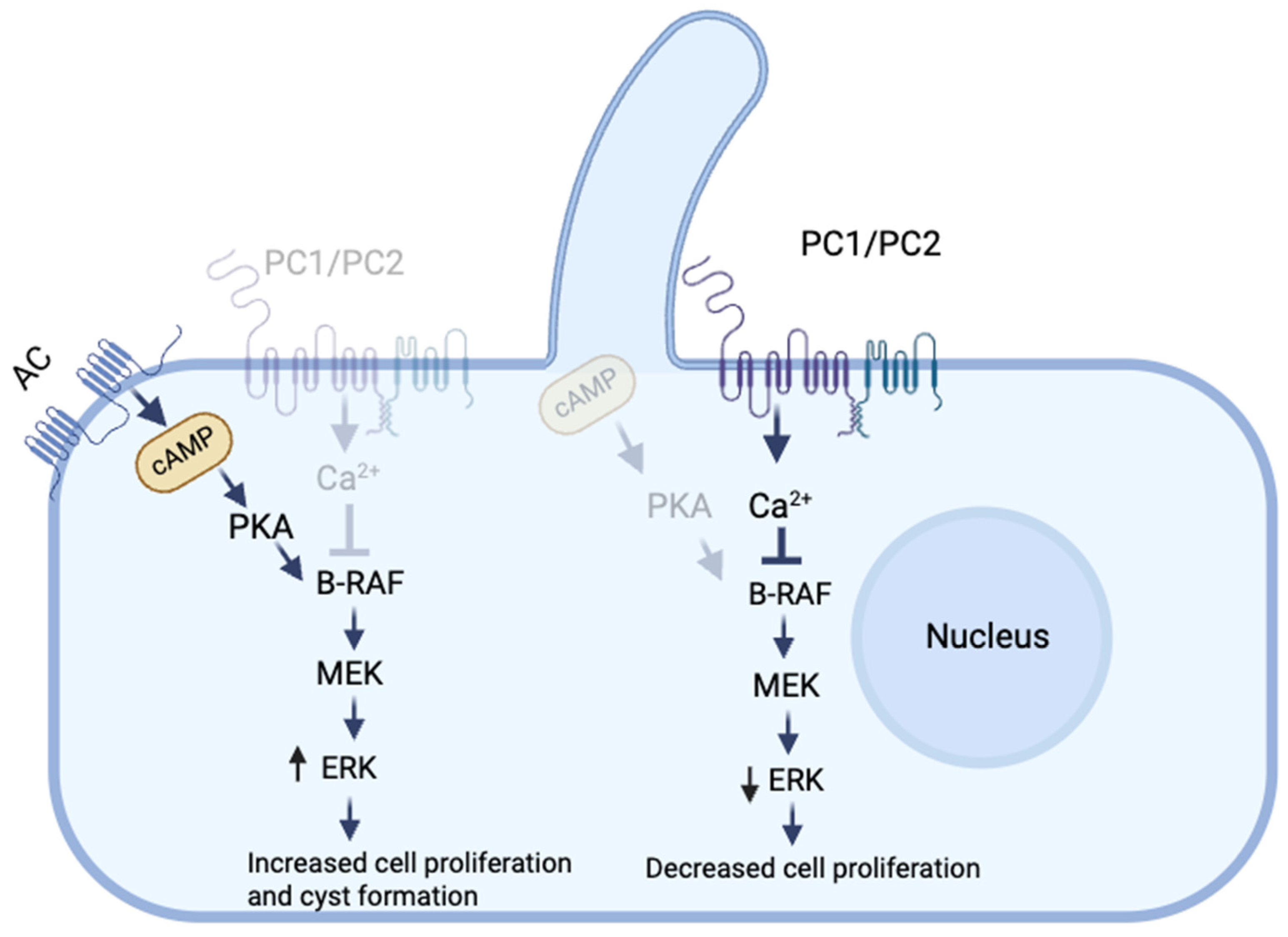
2. Role of Ca+2 in the Pathogenesis of PKD
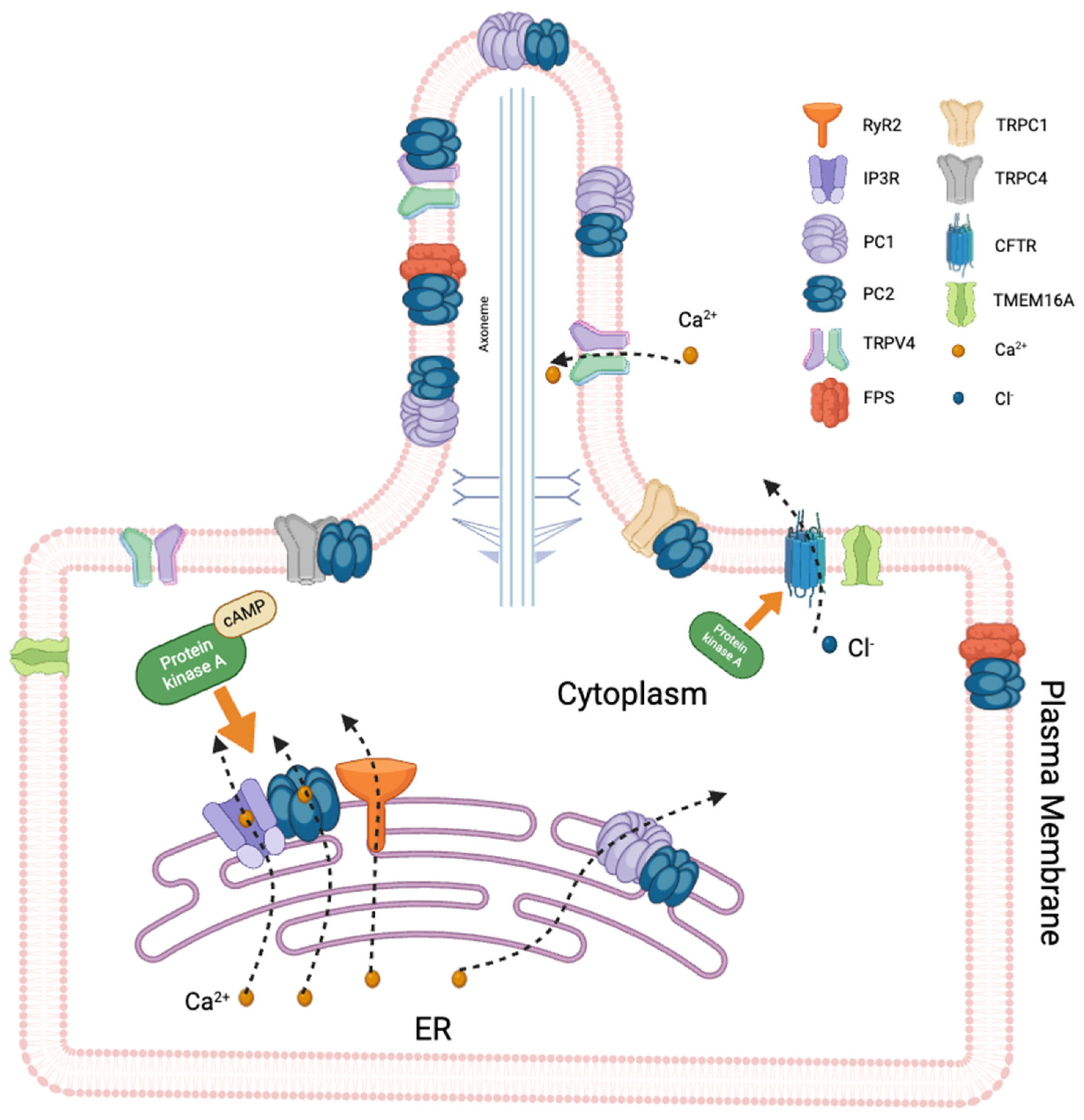
3. Ciliary Ion Channels
3.1. TRP Channels
3.1.1. Polycystins
3.1.2. TRPV4
3.2. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
3.3. Fibrocystin
4. Therapeutic Implications: Targeting Ciliary Ion Channels in PKD
5. Challenges of Studying and Targeting Ciliary Ion Channels
6. Future Directions
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AC | Adenylate cyclase |
| ADPKD | Autosomal dominant polycystic kidney disease |
| ANG II | Angiotensin II |
| ARPKD | Autosomal recessive polycystic kidney disease |
| B-Raf | B-rapidly accelerated fibrosarcoma |
| Ca+2 | Calcium |
| Camp | Cyclic adenosine monophosphate |
| CD | Collecting duct |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| ENAC | Epithelial sodium channel |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ESRD | End-stage renal disease |
| FPC | Fibrocystin |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| MDCK | Polarized Madin–Darby canine kidney |
| MEK | Mitogen-activated protein kinase |
| NCCE | Non-Capacitative Ca+2 Entry |
| NHKS | Normal human kidneys |
| NO | Nitric oxide |
| PC1 | Polycystin-1 |
| PC2 | Polycystin-2 |
| PDEs | Phosphodiesterases |
| PKA | Protein kinase A |
| PKD | Polycystic kidney disease |
| RyR | Ryanodine receptor |
| TMEM16A | Transmembrane member 16A anoctamin 1 |
| TRP | Transient receptor potential |
| TRPA | Transient receptor potential ankyrin |
| TRPC | Transient receptor potential canonical |
| TRPM | Transient receptor potential melastin |
| TRPML | Transient receptor potential mucolipin |
| TRPP | Transient receptor potential polycystin |
| TRPV | Transient receptor potential vanilloid |
| WGS | Whole-genome sequencing |
References
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Foundation, P. Available online: https://pkdcure.org/wp-content/uploads/Welcome-to-PKDF-Information-for-PKDF-Volunteers.pdf (accessed on 27 February 2025).
- Kelleher, C.L.; McFann, K.K.; Johnson, A.M.; Schrier, R.W. Characteristics of hypertension in young adults with autosomal dominant polycystic kidney disease compared with the general US population. Am. J. Hypertens. 2004, 17, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Somlo, S.; Guay-Woodford, L.M. Polycystic kidney disease. In Genetic Diseases of the Kidney; Elsevier: Amsterdam, The Netherlands, 2009; pp. 393–424. [Google Scholar]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.-P.; Hopp, K.; Porath, B.; Shi, B. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J. Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef]
- Besse, W.; Chang, A.R.; Luo, J.Z.; Triffo, W.J.; Moore, B.S.; Gulati, A.; Hartzel, D.N.; Mane, S.; Torres, V.E.; Somlo, S. ALG9 mutation carriers develop kidney and liver cysts. J. Am. Soc. Nephrol. 2019, 30, 2091–2102. [Google Scholar] [CrossRef]
- Senum, S.R.; Li, Y.S.M.; Benson, K.A.; Joli, G.; Olinger, E.; Lavu, S.; Madsen, C.D.; Gregory, A.V.; Neatu, R.; Kline, T.L. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am. J. Hum. Genet. 2022, 109, 136–156. [Google Scholar] [CrossRef]
- Lemoine, H.; Raud, L.; Foulquier, F.; Sayer, J.A.; Lambert, B.; Olinger, E.; Lefèvre, S.; Knebelmann, B.; Harris, P.C.; Trouvé, P. Monoallelic pathogenic ALG5 variants cause atypical polycystic kidney disease and interstitial fibrosis. Am. J. Hum. Genet. 2022, 109, 1484–1499. [Google Scholar] [CrossRef]
- Gabow, P.A. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 1993, 329, 332–342. [Google Scholar] [CrossRef]
- Al-Bhalal, L.; Akhtar, M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD). Adv. Anat. Pathol. 2008, 15, 54–58. [Google Scholar] [CrossRef]
- Blair-Reid, S.A. An Investigation of the Ciliary Protein PKHD1 in Cyst Development in Liver Disease: Clues to the Pathogenesis of Biliary Atresia. Ph.D. Thesis, University of Birmingham, Birmingham, UK, 2010. [Google Scholar]
- Wicher, D.; Obrycki, Ł.; Jankowska, I. Autosomal recessive polycystic kidney disease—The clinical aspects and diagnostic challenges. J. Pediatr. Genet. 2021, 10, 1–8. [Google Scholar] [CrossRef]
- Hoyer, P.F. Clinical manifestations of autosomal recessive polycystic kidney disease. Curr. Opin. Pediatr. 2015, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the kidney: A review. Am. J. Kidney Dis. 2021, 77, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef]
- Fisch, C.; Dupuis-Williams, P. Ultrastructure of cilia and flagella–back to the future! Biol. Cell 2011, 103, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hempson, S.J.; Reif, G.A.; Hedge, A.-M.; Wallace, D.P. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 2006, 17, 178–187. [Google Scholar] [CrossRef]
- Zaika, O.; Mamenko, M.; Berrout, J.; Boukelmoune, N.; O’Neil, R.G.; Pochynyuk, O. TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 604–616. [Google Scholar] [CrossRef]
- Calvet, J.P. The role of calcium and cyclic AMP in PKD. Exon Publ. 2015, 169–196. [Google Scholar]
- Spasic, M.; Jacobs, C.R. Primary cilia: Cell and molecular mechanosensors directing whole tissue function. Proc. Semin. Cell Dev. Biol. 2017, 71, 42–52. [Google Scholar] [CrossRef]
- Lee, K.L.; Guevarra, M.D.; Nguyen, A.M.; Chua, M.C.; Wang, Y.; Jacobs, C.R. The primary cilium functions as a mechanical and calcium signaling nexus. Cilia 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; Indzhykulian, A.; Liu, X.; Li, Y.; Xie, T.; Corey, D.; Clapham, D. Primary cilia are not calcium-responsive mechanosensors. Nature 2016, 531, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Guggino, W.B. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J. Am. Soc. Nephrol. 2000, 11, 1179–1187. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D., Jr.; Pelling, J.C.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994. [Google Scholar] [CrossRef]
- Delmas, P.; Nauli, S.M.; Li, X.; Coste, B.; Osorio, N.; Crest, M.; Brown, D.A.; Zhou, J. Gating of the polycystin ion channel signaling complex in neurons and kidney cells. FASEB J. 2004, 18, 740–742. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Wallace, D.P.; Magenheimer, B.S.; Hempson, S.J.; Grantham, J.J.; Calvet, J.P. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 2004, 279, 40419–40430. [Google Scholar] [CrossRef]
- Pinto, C.S.; Reif, G.A.; Nivens, E.; White, C.; Wallace, D.P. Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl− secretion by human autosomal dominant polycystic kidney cells. Am. J. Physiol. Ren. Physiol. 2012, 303, F1412–F1424. [Google Scholar] [CrossRef]
- Rees, S.; Kittikulsuth, W.; Roos, K.; Strait, K.A.; Van Hoek, A.; Kohan, D.E. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 232–237. [Google Scholar] [CrossRef]
- Saternos, H.; Ley, S.; AbouAlaiwi, W. Primary cilia and calcium signaling interactions. Int. J. Mol. Sci. 2020, 21, 7109. [Google Scholar] [CrossRef]
- Orfali, R.; AlFaiz, A.; Alanazi, M.; Alabdulsalam, R.; Alharbi, M.; Alromaih, Y.; Dallak, I.; Alrahal, M.; Alwatban, A.; Saud, R. TRPV4 Channel Modulators as Potential Drug Candidates for Cystic Fibrosis. Int. J. Mol. Sci. 2024, 25, 10551. [Google Scholar] [CrossRef]
- Tian, P.-F.; Sun, M.-M.; Hu, X.-Y.; Du, J.; He, W. TRPP2 ion channels: The roles in various subcellular locations. Biochimie 2022, 201, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Sudarikova, A.V.; Vasileva, V.Y.; Sultanova, R.F.; Ilatovskaya, D.V. Recent advances in understanding ion transport mechanisms in polycystic kidney disease. Clin. Sci. 2021, 135, 2521–2540. [Google Scholar] [CrossRef] [PubMed]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef]
- Snyder, D.S.; Tradtrantip, L.; Yao, C.; Kurth, M.J.; Verkman, A. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J. Med. Chem. 2011, 54, 5468–5477. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Sonawane, N.; Namkung, W.; Verkman, A. Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J. Med. Chem. 2009, 52, 6447–6455. [Google Scholar] [CrossRef]
- Su, L.; Yuan, H.; Zhang, H.; Wang, R.; Fu, K.; Yin, L.; Ren, Y.; Liu, H.; Fang, Q.; Wang, J. PF–06409577 inhibits renal cyst progression by concurrently inhibiting the mTOR pathway and CFTR channel activity. FEBS Open Bio 2022, 12, 1761–1770. [Google Scholar] [CrossRef]
- Enyedi, P.; Czirják, G. Molecular background of leak K+ currents: Two-pore domain potassium channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef]
- Hofherr, A.; Köttgen, M. TRPP channels and polycystins. Transient Recept. Potential Channels 2011, 287–313. [Google Scholar]
- Lemos, F.O.; Ehrlich, B.E. Polycystin and calcium signaling in cell death and survival. Cell Calcium 2018, 69, 37–45. [Google Scholar] [CrossRef]
- Parnell, S.C.; Magenheimer, B.S.; Maser, R.L.; Pavlov, T.S.; Havens, M.A.; Hastings, M.L.; Jackson, S.F.; Ward, C.J.; Peterson, K.R.; Staruschenko, A. A mutation affecting polycystin-1 mediated heterotrimeric G-protein signaling causes PKD. Hum. Mol. Genet. 2018, 27, 3313–3324. [Google Scholar] [CrossRef]
- Parnell, S.C.; Magenheimer, B.S.; Maser, R.L.; Rankin, C.A.; Smine, A.; Okamoto, T.; Calvet, J.P. The Polycystic Kidney Disease-1 Protein, Polycystin-1, Binds and Activates Heterotrimeric G-Proteinsin Vitro. Biochem. Biophys. Res. Commun. 1998, 251, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.L.; Calvet, J.P.; Parnell, S.C. The GPCR properties of polycystin-1-A new paradigm. Front. Mol. Biosci. 2022, 9, 1035507. [Google Scholar] [CrossRef] [PubMed]
- Grieben, M.; Pike, A.C.; Shintre, C.A.; Venturi, E.; El-Ajouz, S.; Tessitore, A.; Shrestha, L.; Mukhopadhyay, S.; Mahajan, P.; Chalk, R. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 2017, 24, 114–122. [Google Scholar] [CrossRef]
- Newby, L.J.; Streets, A.J.; Zhao, Y.; Harris, P.C.; Ward, C.J.; Ong, A.C. Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J. Biol. Chem. 2002, 277, 20763–20773. [Google Scholar] [CrossRef]
- Ha, K.; Nobuhara, M.; Wang, Q.; Walker, R.V.; Qian, F.; Schartner, C.; Cao, E.; Delling, M. The heteromeric PC-1/PC-2 polycystin complex is activated by the PC-1 N-terminus. eLife 2020, 9, e60684. [Google Scholar] [CrossRef]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and-2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [CrossRef]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, eaat9819. [Google Scholar] [CrossRef]
- Santoso, N.G.; Cebotaru, L.; Guggino, W.B. Polycystin-1, 2, and STIM1 interact with IP3R to modulate ER Ca2+ release through the PI3K/Akt pathway. Cell. Physiol. Biochem. 2011, 27, 715–726. [Google Scholar] [CrossRef]
- Aguiari, G.; Campanella, M.; Manzati, E.; Pinton, P.; Banzi, M.; Moretti, S.; Piva, R.; Rizzuto, R.; del Senno, L. Expression of polycystin-1 C-terminal fragment enhances the ATP-induced Ca2+ release in human kidney cells. Biochem. Biophys. Res. Commun. 2003, 301, 657–664. [Google Scholar] [CrossRef]
- Manzati, E.; Aguiari, G.; Banzi, M.; Manzati, M.; Selvatici, R.; Falzarano, S.; Maestri, I.; Pinton, P.; Rizzuto, R.; del Senno, L. The cytoplasmic C-terminus of polycystin-1 increases cell proliferation in kidney epithelial cells through serum-activated and Ca2+-dependent pathway(s). Exp. Cell Res. 2005, 304, 391–406. [Google Scholar] [CrossRef]
- Puri, S.; Magenheimer, B.S.; Maser, R.L.; Ryan, E.M.; Zien, C.A.; Walker, D.D.; Wallace, D.P.; Hempson, S.J.; Calvet, J.P. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J. Biol. Chem. 2004, 279, 55455–55464. [Google Scholar] [CrossRef] [PubMed]
- Aguiari, G.; Trimi, V.; Bogo, M.; Mangolini, A.; Szabadkai, G.; Pinton, P.; Witzgall, R.; Harris, P.; Borea, P.A.; Rizzuto, R. Novel role for polycystin-1 in modulating cell proliferation through calcium oscillations in kidney cells. Cell Prolif. 2008, 41, 554–573. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Vien, T.; Duan, J.; Sheu, S.-H.; DeCaen, P.G.; Clapham, D.E. Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. eLife 2018, 7, e33183. [Google Scholar] [CrossRef] [PubMed]
- Anyatonwu, G.I.; Estrada, M.; Tian, X.; Somlo, S.; Ehrlich, B.E. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc. Natl. Acad. Sci. USA 2007, 104, 6454–6459. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef]
- Li, Y.; Wright, J.M.; Qian, F.; Germino, G.G.; Guggino, W.B. Polycystin 2 interacts with type I inositol 1, 4, 5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J. Biol. Chem. 2005, 280, 41298–41306. [Google Scholar] [CrossRef]
- Abdi, A.; Mazzocco, C.; Légeron, F.-P.; Yvert, B.; Macrez, N.; Morel, J.-L. TRPP2 modulates ryanodine-and inositol-1, 4, 5-trisphosphate receptors-dependent Ca2+ signals in opposite ways in cerebral arteries. Cell Calcium 2015, 58, 467–475. [Google Scholar] [CrossRef]
- Park, S.K.; Kim, K.; Page, G.P.; Allison, D.B.; Weindruch, R.; Prolla, T.A. Gene expression profiling of aging in multiple mouse strains: Identification of aging biomarkers and impact of dietary antioxidants. Aging Cell 2009, 8, 484–495. [Google Scholar] [CrossRef]
- Ungvari, Z.; Kaley, G.; De Cabo, R.; Sonntag, W.E.; Csiszar, A. Mechanisms of vascular aging: New perspectives. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2010, 65, 1028–1041. [Google Scholar] [CrossRef]
- AbouAlaiwi, W.A.; Takahashi, M.; Mell, B.R.; Jones, T.J.; Ratnam, S.; Kolb, R.J.; Nauli, S.M. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 2009, 104, 860–869. [Google Scholar] [CrossRef]
- Tsiokas, L.; Arnould, T.; Zhu, C.; Kim, E.; Walz, G.; Sukhatme, V.P. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc. Natl. Acad. Sci. USA 1999, 96, 3934–3939. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ding, M.; Sours-Brothers, S.; Graham, S.; Ma, R. Mediation of angiotensin II-induced Ca2+ signaling by polycystin 2 in glomerular mesangial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F909–F918. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.L.; Ehrlich, B.E. Polycystin 2: A calcium channel, channel partner, and regulator of calcium homeostasis in ADPKD. Cell. Signal. 2020, 66, 109490. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yu, S. New insights into the molecular mechanisms targeting tubular channels/transporters in PKD development. Kidney Dis. 2016, 2, 128–135. [Google Scholar] [CrossRef]
- Deng, X.; Wang, Y.; Zhou, Y.; Soboloff, J.; Gill, D.L. STIM and Orai: Dynamic intermembrane coupling to control cellular calcium signals. J. Biol. Chem. 2009, 284, 22501–22505. [Google Scholar] [CrossRef]
- Yanda, M.K.; Liu, Q.; Cebotaru, V.; Guggino, W.B.; Cebotaru, L. Role of calcium in adult onset polycystic kidney disease. Cell. Signal. 2019, 53, 140–150. [Google Scholar] [CrossRef]
- Kottgen, M.; Buchholz, B.R.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef]
- Voets, T.; Prenen, J.; Vriens, J.; Watanabe, H.; Janssens, A.; Wissenbach, U.; Bo, M.; Droogmans, G.; Nilius, B. Molecular determinants of permeation through the cation channel TRPV4. J. Biol. Chem. 2002, 277, 33704–33710. [Google Scholar] [CrossRef]
- Tomilin, V.; Reif, G.A.; Zaika, O.; Wallace, D.P.; Pochynyuk, O. Deficient transient receptor potential vanilloid type 4 function contributes to compromised [Ca2+] i homeostasis in human autosomal-dominant polycystic kidney disease cells. FASEB J. 2018, 32, 4612. [Google Scholar] [CrossRef]
- Mizuno, A.; Matsumoto, N.; Imai, M.; Suzuki, M. Impaired osmotic sensation in mice lacking TRPV4. Am. J. Physiol. Cell Physiol. 2003, 285, C96–C101. [Google Scholar] [CrossRef]
- Taniguchi, J.; Tsuruoka, S.; Mizuno, A.; Sato, J.-I.; Fujimura, A.; Suzuki, M. TRPV4 as a flow sensor in flow-dependent K+ secretion from the cortical collecting duct. Am. J. Physiol. Ren. Physiol. 2007, 292, F667–F673. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, S.D.; Ramjeesingh, M.; Bear, C.E.; Argent, B.E.; Linsdell, P.; Gray, M.A. The cystic fibrosis transmembrane conductance regulator is an extracellular chloride sensor. Pflug. Arch. 2015, 467, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Yanda, M.K.; Ciobanu, C.; Guggino, W.B.; Cebotaru, L. CFTR and PC2, partners in the primary cilia in autosomal dominant polycystic kidney disease. Am. J. Physiol. Cell Physiol. 2023, 325, C682–C693. [Google Scholar] [CrossRef]
- Hanaoka, K.; Devuyst, O.; Schwiebert, E.M.; Wilson, P.D.; Guggino, W.B. A role for CFTR in human autosomal dominant polycystic kidney disease. Am. J. Physiol. 1996, 270, C389–C399. [Google Scholar] [CrossRef]
- Vasileva, V.Y.; Sultanova, R.F.; Sudarikova, A.V.; Ilatovskaya, D.V. Insights Into the Molecular Mechanisms of Polycystic Kidney Diseases. Front. Physiol. 2021, 12, 693130. [Google Scholar] [CrossRef]
- Lebeau, C.; Hanaoka, K.; Moore-Hoon, M.L.; Guggino, W.B.; Beauwens, R.; Devuyst, O. Basolateral chloride transporters in autosomal dominant polycystic kidney disease. Pflügers Arch. 2002, 444, 722–731. [Google Scholar] [CrossRef]
- Jouret, F.; Devuyst, O. Targeting chloride transport in autosomal dominant polycystic kidney disease. Cell. Signal. 2020, 73, 109703. [Google Scholar] [CrossRef]
- Yanda, M.K.; Cha, B.; Cebotaru, C.V.; Cebotaru, L. Pharmacological reversal of renal cysts from secretion to absorption suggests a potential therapeutic strategy for managing autosomal dominant polycystic kidney disease. J. Biol. Chem. 2019, 294, 17090–17104. [Google Scholar] [CrossRef]
- Yang, B.; Sonawane, N.D.; Zhao, D.; Somlo, S.; Verkman, A. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 1300–1310. [Google Scholar] [CrossRef]
- Sullivan, L.P.; Wallace, D.P.; Grantham, J.J. Epithelial transport in polycystic kidney disease. Physiol. Rev. 1998, 78, 1165–1191. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, R.; Ousingsawat, J.; Cabrita, I.; Pinto, M.; Lérias, J.R.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Plasma membrane–localized TMEM16 proteins are indispensable for expression of CFTR. J. Mol. Med. 2019, 97, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Faria, D.; Schley, G.; Schreiber, R.; Eckardt, K.-U.; Kunzelmann, K. Anoctamin 1 induces calcium-activated chloride secretion and proliferation of renal cyst–forming epithelial cells. Kidney Int. 2014, 85, 1058–1067. [Google Scholar] [CrossRef]
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat. Commun. 2020, 11, 4320. [Google Scholar] [CrossRef]
- Wang, S.; Luo, Y.; Wilson, P.D.; Witman, G.B.; Zhou, J. The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J. Am. Soc. Nephrol. 2004, 15, 592–602. [Google Scholar] [CrossRef]
- Ward, C.J.; Yuan, D.; Masyuk, T.V.; Wang, X.; Punyashthiti, R.; Whelan, S.; Bacallao, R.; Torra, R.; LaRusso, N.F.; Torres, V.E. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003, 12, 2703–2710. [Google Scholar] [CrossRef]
- Zhang, M.-Z.; Mai, W.; Li, C.; Cho, S.-Y.; Hao, C.; Moeckel, G.; Zhao, R.; Kim, I.; Wang, J.; Xiong, H. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2311–2316. [Google Scholar] [CrossRef]
- Kim, I.; Fu, Y.; Hui, K.; Moeckel, G.; Mai, W.; Li, C.; Liang, D.; Zhao, P.; Ma, J.; Chen, X.-Z. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J. Am. Soc. Nephrol. 2008, 19, 455–468. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Nauli, S.M.; Li, X.; Starremans, P.G.; Luo, Y.; Roberts, K.A.; Zhou, J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol. Cell. Biol. 2007, 27, 3241–3252. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Zhang, S.; Zhou, Q.; Guo, H.; Zhang, K.; Zheng, R.; Xiao, C. PKHD1 gene silencing may cause cell abnormal proliferation through modulation of intracellular calcium in autosomal recessive polycystic kidney disease. BMB Rep. 2007, 40, 467–474. [Google Scholar] [CrossRef]
- Wu, Y.; Dai, X.-Q.; Li, Q.; Chen, C.X.; Mai, W.; Hussain, Z.; Long, W.; Montalbetti, N.; Li, G.; Glynne, R. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum. Mol. Genet. 2006, 15, 3280–3292. [Google Scholar] [CrossRef] [PubMed]
- Leuenroth, S.J.; Okuhara, D.; Shotwell, J.D.; Markowitz, G.S.; Yu, Z.; Somlo, S.; Crews, C.M. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2007, 104, 4389–4394. [Google Scholar] [CrossRef] [PubMed]
- Gattone, V.H.; Chen, N.X.; Sinders, R.M.; Seifert, M.F.; Duan, D.; Martin, D.; Henley, C.; Moe, S.M. Calcimimetic inhibits late-stage cyst growth in ADPKD. J. Am. Soc. Nephrol. 2009, 20, 1527–1532. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Masyuk, T.V.; Huang, B.Q.; Banales, J.M.; Lehmann, G.L.; Radtke, B.N.; Stroope, A.; Masyuk, A.I.; Splinter, P.L.; LaRusso, N.F. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology 2010, 139, 304–314.e302. [Google Scholar] [CrossRef]
- Seo, Y.; Kim, J.; Chang, J.; Kim, S.S.; Namkung, W.; Kim, I. Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur. J. Med. Chem. 2018, 160, 245–255. [Google Scholar] [CrossRef]
- Yanda, M.K.; Liu, Q.; Cebotaru, L. A potential strategy for reducing cysts in autosomal dominant polycystic kidney disease with a CFTR corrector. J. Biol. Chem. 2018, 293, 11513–11526. [Google Scholar] [CrossRef]
- Yuajit, C.; Homvisasevongsa, S.; Chatsudthipong, L.; Soodvilai, S.; Muanprasat, C.; Chatsudthipong, V. Steviol reduces MDCK Cyst formation and growth by inhibiting CFTR channel activity and promoting proteasome-mediated CFTR degradation. PLoS ONE 2013, 8, e58871. [Google Scholar] [CrossRef]
- Wang, J.; Tripathy, N.; Chung, E.J. Targeting and therapeutic peptide-based strategies for polycystic kidney disease. Adv. Drug Deliv. Rev. 2020, 161, 176–189. [Google Scholar] [CrossRef]
- He, S.; Chen, M.; Lin, X.; Lv, Z.; Liang, R.; Huang, L. Triptolide inhibits PDGF-induced proliferation of ASMCs through G0/G1 cell cycle arrest and suppression of the AKT/NF-κB/cyclinD1 signaling pathway. Eur. J. Pharmacol. 2020, 867, 172811. [Google Scholar] [CrossRef]
- Koulen, P.; Cai, Y.; Geng, L.; Maeda, Y.; Nishimura, S.; Witzgall, R.; Ehrlich, B.E.; Somlo, S. Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 2002, 4, 191–197. [Google Scholar] [CrossRef]
- Guay-Woodford, L.M.; Henske, E.; Igarashi, P.; Perrone, R.D.; Reed-Gitomer, B.; Somlo, S.; Torres, V.E.; Ketchum, C.J.; Star, R.A.; Flessner, M.F. Filling the holes in cystic kidney disease research. Clin. J. Am. Soc. Nephrol. 2014, 9, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Pablo, J.L.; DeCaen, P.G.; Clapham, D.E. Progress in ciliary ion channel physiology. J. Gen. Physiol. 2017, 149, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.A.; Szabo, G.; Otero, A.D.S. Methods for the isolation of sensory and primary cilia: An overview. Methods Cell Biol. 2009, 94, 87–101. [Google Scholar]
- Pala, R.; Mohieldin, A.M.; Sherpa, R.T.; Kathem, S.H.; Shamloo, K.; Luan, Z.; Zhou, J.; Zheng, J.-G.; Ahsan, A.; Nauli, S.M. Ciliotherapy: Remote control of primary cilia movement and function by magnetic nanoparticles. ACS Nano 2019, 13, 3555–3572. [Google Scholar] [CrossRef]
- Pala, R.; Nauli, S.M. Targeting primary cilia with polymeric nanoparticles. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Wang, S.; Kang, Y.; Xie, H. PKD2: An Important Membrane Protein in Organ Development. Cells 2024, 13, 1722. [Google Scholar] [CrossRef]
- Bais, T.; Gansevoort, R.T.; Meijer, E. Drugs in clinical development to treat autosomal dominant polycystic kidney disease. Drugs 2022, 82, 1095–1115. [Google Scholar] [CrossRef]
- Cruz, N.M.; Freedman, B.S. CRISPR gene editing in the kidney. Am. J. Kidney Dis. 2018, 71, 874–883. [Google Scholar] [CrossRef]
- Vishy, C.E.; Thomas, C.; Vincent, T.; Crawford, D.K.; Goddeeris, M.M.; Freedman, B.S. Genetics of cystogenesis in base-edited human organoids reveal therapeutic strategies for polycystic kidney disease. Cell Stem Cell 2024, 31, 537–553.e535. [Google Scholar] [CrossRef]
- Gómez-García, F.; Martínez-Pulleiro, R.; Carrera, N.; Allegue, C.; Garcia-Gonzalez, M.A. Genetic kidney diseases (GKDs) modeling using genome editing technologies. Cells 2022, 11, 1571. [Google Scholar] [CrossRef]
- Hunt, J.M.T.; Samson, C.A.; Rand, A.D.; Sheppard, H.M. Unintended CRISPR-Cas9 editing outcomes: A review of the detection and prevalence of structural variants generated by gene-editing in human cells. Hum. Genet. 2023, 142, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Wang, C.; Chen, L.; Shen, W.; Xie, Y.; Li, W.; Li, Q. Novel method for the genomic analysis of PKD1 mutation in autosomal dominant polycystic kidney disease. Front. Cell Dev. Biol. 2023, 10, 937580. [Google Scholar] [CrossRef] [PubMed]
- Allemailem, K.S.; Alsahli, M.A.; Almatroudi, A.; Alrumaihi, F.; Alkhaleefah, F.K.; Rahmani, A.H.; Khan, A.A. Current updates of CRISPR/Cas9–mediated genome editing and targeting within tumor cells: An innovative strategy of cancer management. Cancer Commun. 2022, 42, 1257–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, L.; Jiang, J.; Wu, M.; Lin, P. Applications and challenges of CRISPR-Cas gene-editing to disease treatment in clinics. Precis. Clin. Med. 2021, 4, 179–191. [Google Scholar] [CrossRef]
- McIntyre, J.C.; Williams, C.L.; Martens, J.R. Smelling the roses and seeing the light: Gene therapy for ciliopathies. Trends Biotechnol. 2013, 31, 355–363. [Google Scholar] [CrossRef]
- Peek, J.L.; Wilson, M.H. Cell and gene therapy for kidney disease. Nat. Rev. Nephrol. 2023, 19, 451–462. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Drug | Target and MOA | Pre-Clinical
Outcome | Clinical Trial Outcome |
|---|---|---|---|
| Triptolide | Induces Ca+2 release through a PC2-dependent pathway [95] Cell cycle arrest in G0 phase [102] | Triptolide was shown in experimental studies to inhibit cyst formation and growth [95] | Triptolide demonstrated effectiveness in managing PKD, as evidenced by its association with a significant reduction in proteinuria in PKD patients during an uncontrolled clinical trial. However, the volume of polycystic kidney and eGFR were not influenced [102] |
| R-568 | Calcium-sensing receptor | R-568 inhibited cyst growth and fibrosis in late stage PKD | - |
| 4αPDD and GSK1016790 | TRPV4 activator | TRPV4 activation in PKD cholangiocytes increased [Ca+2]i by 30%, inhibiting cell proliferation by approximately 25–50% and cyst growth in 3-dimensional cultures (3-fold) [97] | - |
| GSK1016790A | TRPV4 activator | Long-term systemic treatment with GSK1016790A significantly reduced the renal manifestations of PKD in a time-dependent manner. At the cellular level, GSK1016790A restored mechanosensitive Ca+2 signaling and improved both the function and the subcellular distribution of TRPV4 [21] | - |
| Niclosamide and Benzbromarone | Non-selective TMEM16A inhibitors. Block Cl− currents and inhibit the expression of TMEM16A upon long-term treatment | The knockout of TMEM16A or the inhibition of TMEM16A in vivo using niclosamide and benzbromarone, significantly reduced cyst growth and abnormal cell proliferation [87] | - |
| Ani9 | Selective TMEM16A inhibitors [98] | Ani9 significantly reduced cyst growth and abnormal cell proliferation in a PKD mouse model [87] | - |
| VX-809 | Corrector of CFTR; VX-809 down-regulates cAMP levels, which reduced cell proliferation. It also affects adenylyl cyclase 3 activity, leading to a decrease in resting intracellular Ca+2 levels and the release of Ca+2 from the endoplasmic reticulum [99] | VX-809 at 30 mg/kg to mice or at 10 μm to cells did significantly inhibit cell proliferation when compared with control mice or cells [99] | - |
| Steviol | Inhibiting CFTR chloride channel activity and reducing CFTR expression [100] | Steviol inhibited the forskolin-stimulated apical chloride current in MDCK epithelium in a dose-dependent manner. Prolonged treatment with 100 µM steviol for 24 h significantly reduced this chloride current, partially by decreasing CFTR protein expression in MDCK cells [100] | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshriem, L.A.; Buqaileh, R.; Alorjani, Q.; AbouAlaiwi, W. Ciliary Ion Channels in Polycystic Kidney Disease. Cells 2025, 14, 459. https://doi.org/10.3390/cells14060459
Alshriem LA, Buqaileh R, Alorjani Q, AbouAlaiwi W. Ciliary Ion Channels in Polycystic Kidney Disease. Cells. 2025; 14(6):459. https://doi.org/10.3390/cells14060459
Chicago/Turabian StyleAlshriem, Lubna A., Raghad Buqaileh, Qasim Alorjani, and Wissam AbouAlaiwi. 2025. "Ciliary Ion Channels in Polycystic Kidney Disease" Cells 14, no. 6: 459. https://doi.org/10.3390/cells14060459
APA StyleAlshriem, L. A., Buqaileh, R., Alorjani, Q., & AbouAlaiwi, W. (2025). Ciliary Ion Channels in Polycystic Kidney Disease. Cells, 14(6), 459. https://doi.org/10.3390/cells14060459