
Interleukin 24 Promotes Mitochondrial Dysfunction, Glucose Regulation, and Apoptosis by Inactivating Glycogen Synthase Kinase 3 Beta in Human Prostate Cancer Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Virus Infection
2.3. Plasmids and Transfections
2.4. Western Blot Analysis
2.5. Viability Assays
2.6. Annexin V Binding Assays
2.7. Mitochondrial Membrane Potential
2.8. GSK3β Kinase Assay
2.9. Intracellular Glucose Measurement
2.10. Statistical Analysis
3. Results
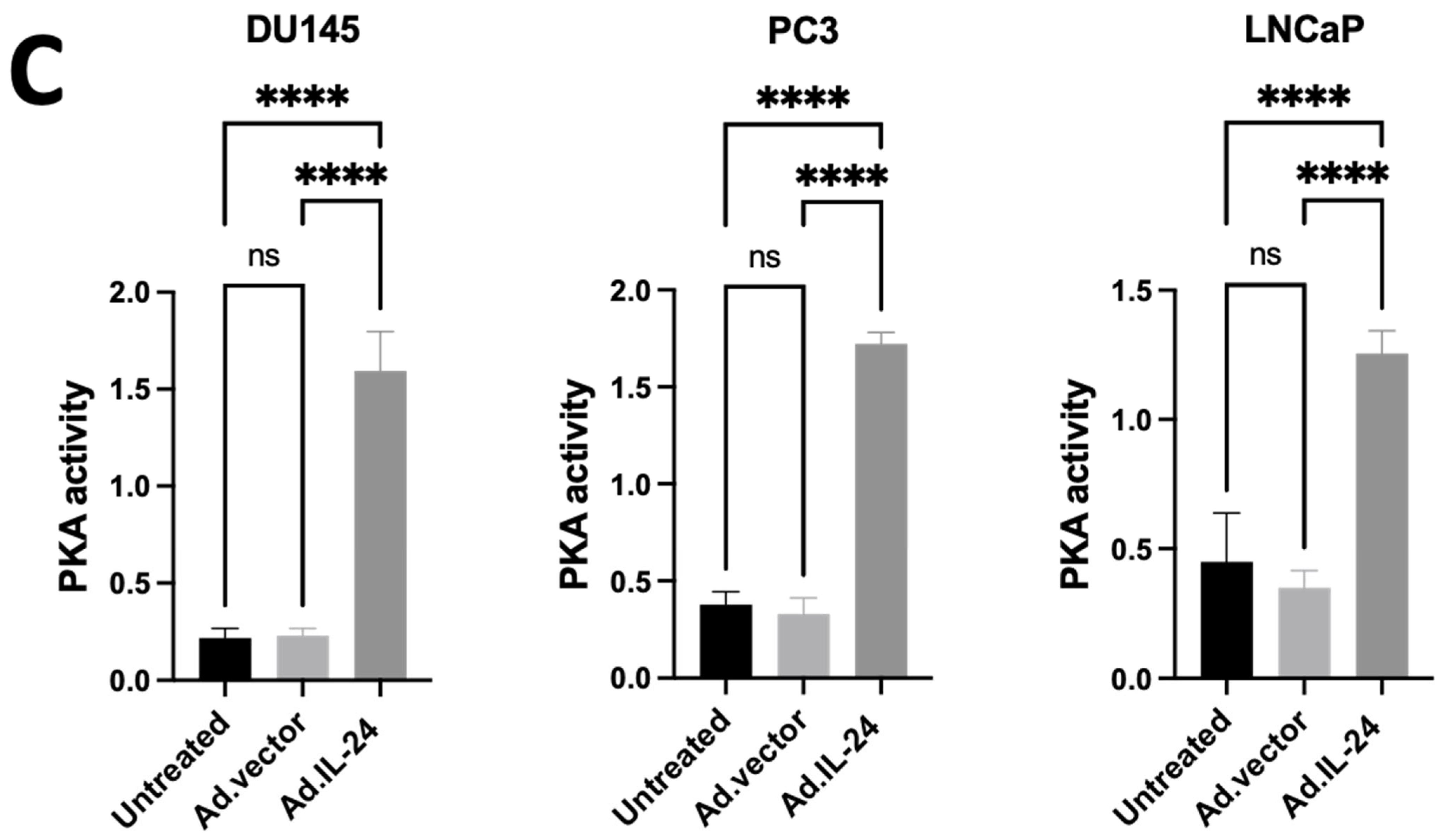
3.1. IL-24-Mediated Activation of PKA in Human Prostate Cancer Cells
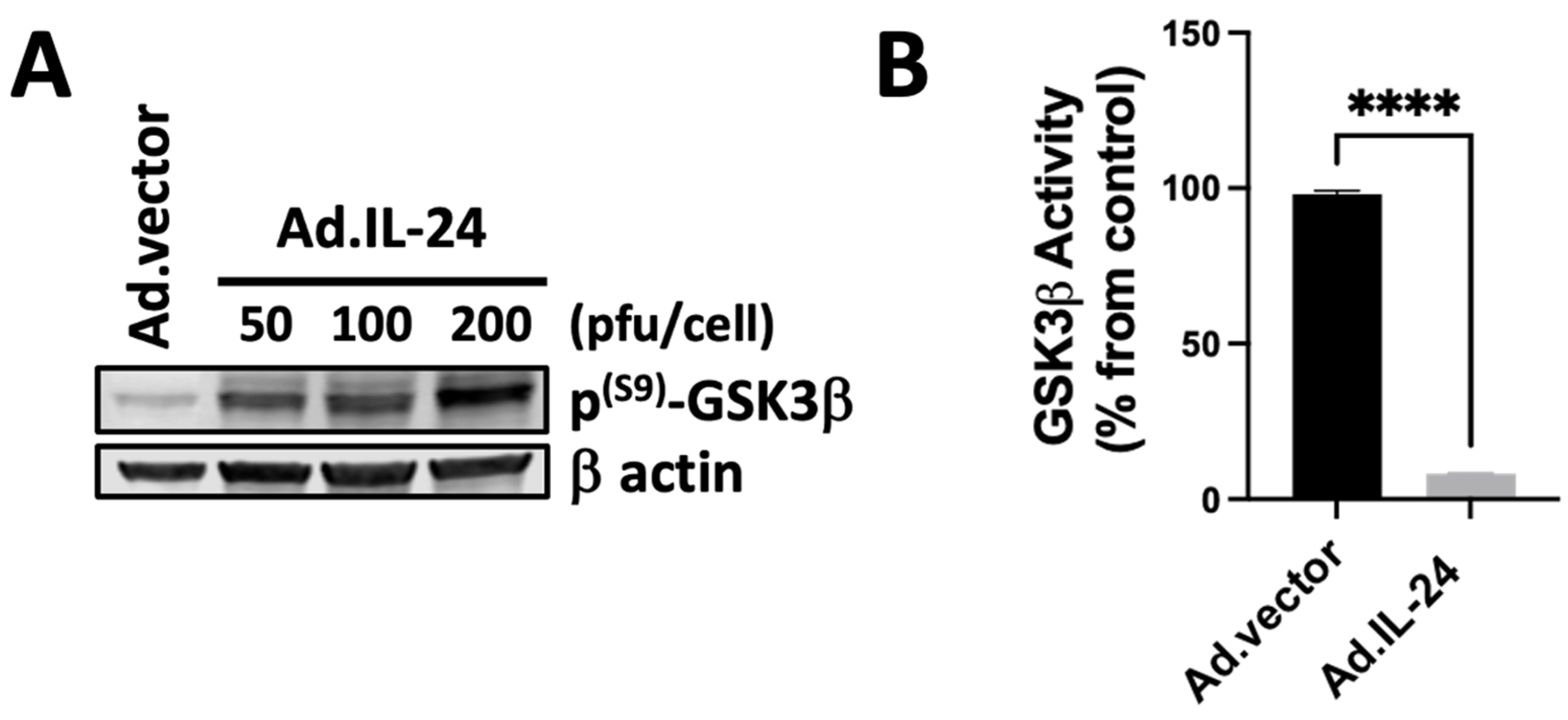
3.2. IL-24-Mediated Inactivation of GSK3β Kinase
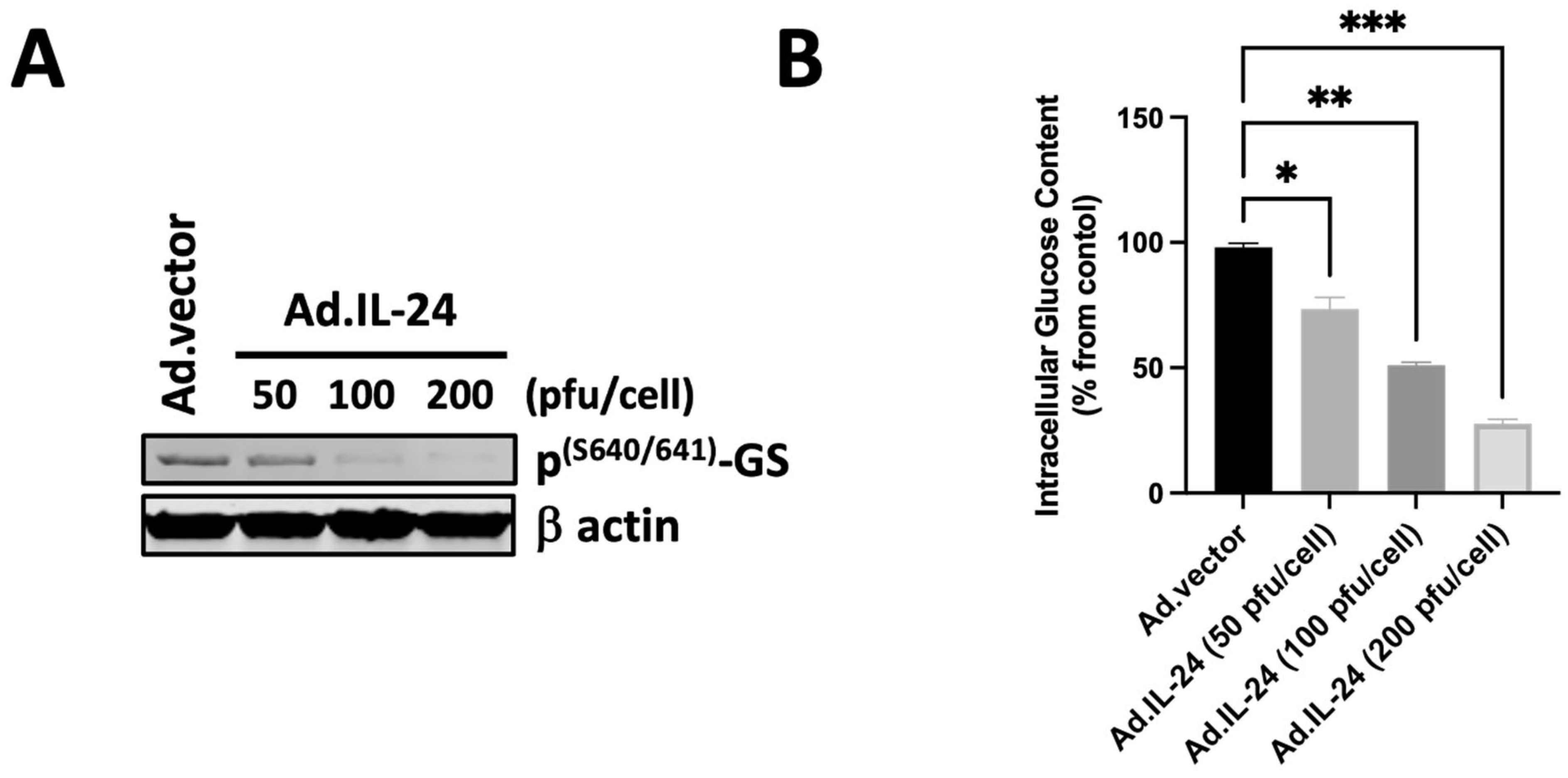
3.3. IL-24-Mediated Activation of Glycogen Synthase and Its Effect on Glucose Levels
3.4. IL-24-Mediated Inactivation of GSK3β Kinase in Human Prostate Cancer Cells via PKA
3.5. IL-24-Dependent Phosphorylation of GSK3β Is Necessary to Mediate Apoptosis in Human Prostate Cancer Cells
3.6. IL-24-Dependent Phosphorylation of GSK3β Inhibits Anti-Apoptotic Mechanisms in Mitochondria
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emdad, L.; Bhoopathi, P.; Talukdar, S.; Pradhan, A.K.; Sarkar, D.; Wang, X.-Y.; Das, S.K.; Fisher, P.B. Recent Insights into Apoptosis and Toxic Autophagy: The Roles of MDA-7/IL-24, a Multidimensional Anti-Cancer Therapeutic. Semin. Cancer Biol. 2020, 66, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Lopez, S.; Kim, A.; Kasteri, J.; Olumuyide, E.; Punu, K.; de la Parra, C.; Sauane, M. Interleukin 24: Signal Transduction Pathways. Cancers 2023, 15, 3365. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Bhutia, S.K.; Azab, B.; Su, Z.; Quinn, B.A.; Kegelmen, T.P.; Das, S.K.; Kim, K.; Lee, S.-G.; Park, M.A.; et al. Mda-7/IL-24: A Unique Member of the IL-10 Gene Family Promoting Cancer-Targeted Toxicity. Cytokine Growth Factor Rev. 2010, 21, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Garn, H.; Schmidt, A.; Grau, V.; Stumpf, S.; Kaufmann, A.; Becker, M.; Gemsa, D.; Siese, A. IL-24 Is Expressed by Rat and Human Macrophages. Immunobiology 2002, 205, 321–334. [Google Scholar] [CrossRef]
- Liu, S.; Hur, Y.H.; Cai, X.; Cong, Q.; Yang, Y.; Xu, C.; Bilate, A.M.; Gonzales, K.A.U.; Parigi, S.M.; Cowley, C.J.; et al. A Tissue Injury Sensing and Repair Pathway Distinct from Host Pathogen Defense. Cell 2023, 186, 2127–2143.e22. [Google Scholar] [CrossRef]
- Sauane, M.; Gopalkrishnan, R.V.; Sarkar, D.; Su, Z.Z.; Lebedeva, I.V.; Dent, P.; Pestka, S.; Fisher, P.B. MDA-7/IL-24: Novel Cancer Growth Suppressing and Apoptosis Inducing Cytokine. Cytokine Growth Factor Rev. 2003, 14, 35–51. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, X.; Chong, W. Interleukin-24 Immunobiology and Its Roles in Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 627. [Google Scholar] [CrossRef]
- Turchin, I.; Bourcier, M. The Role of Interleukins in the Pathogenesis of Dermatological Immune-Mediated Diseases. Adv. Ther. 2022, 39, 4474–4508. [Google Scholar] [CrossRef]
- Xu, X.; Prens, E.; Florencia, E.; Leenen, P.; Boon, L.; Asmawidjaja, P.; Mus, A.-M.; Lubberts, E. Interleukin-17A Drives IL-19 and IL-24 Expression in Skin Stromal Cells Regulating Keratinocyte Proliferation. Front. Immunol. 2021, 12, 719562. [Google Scholar] [CrossRef]
- Qian, X.; Tong, M.; Zhang, T.; Li, Q.; Hua, M.; Zhou, N.; Zeng, W. IL-24 Promotes Atopic Dermatitis-like Inflammation through Driving MRSA-Induced Allergic Responses. Protein Cell 2024, XX, pwae030. [Google Scholar] [CrossRef]
- Sahoo, A.; Im, S.-H. Molecular Mechanisms Governing IL-24 Gene Expression. Immune Netw. 2012, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Modi, J.; Roy, A.; Pradhan, A.K.; Kumar, A.; Talukdar, S.; Bhoopathi, P.; Maji, S.; Mannangatti, P.; De La Rosa, D.S.; Li, J.; et al. Insights into the Mechanisms of Action of MDA-7/IL-24: A Ubiquitous Cancer-Suppressing Protein. Int. J. Mol. Sci. 2021, 23, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Xu, Y. Interleukin-24 Regulates T Cell Activity in Patients With Colorectal Adenocarcinoma. Front. Oncol. 2019, 9, 1401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lv, J.; Zhang, T. Combination of IL-24 and Cisplatin Inhibits Angiogenesis and Lymphangiogenesis of Cervical Cancer Xenografts in a Nude Mouse Model by Inhibiting VEGF, VEGF-C and PDGF-B. Oncol. Rep. 2015, 33, 2468–2476. [Google Scholar] [CrossRef]
- Zhuo, B.; Shi, Y.; Qin, H.; Sun, Q.; Li, Z.; Zhang, F.; Wang, R.; Wang, X. Interleukin-24 Inhibits Osteosarcoma Cell Migration and Invasion via the JNK/c-Jun Signaling Pathways. Oncol. Lett. 2017, 13, 4505–4511. [Google Scholar] [CrossRef]
- Panneerselvam, J.; Srivastava, A.; Muralidharan, R.; Wang, Q.; Zheng, W.; Zhao, L.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. IL-24 Modulates the High Mobility Group (HMG) A1/miR222 /AKT Signaling in Lung Cancer Cells. Oncotarget 2016, 7, 70247–70263. [Google Scholar] [CrossRef]
- Persaud, L.; Mighty, J.; Zhong, X.; Francis, A.; Mendez, M.; Muharam, H.; Redenti, S.M.; Das, D.; Aktas, B.H.; Sauane, M. IL-24 Promotes Apoptosis through cAMP-Dependent PKA Pathways in Human Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3561. [Google Scholar] [CrossRef]
- Zhong, X.; Persaud, L.; Muharam, H.; Francis, A.; Das, D.; Aktas, B.H.; Sauane, M. Eukaryotic Translation Initiation Factor 4A Down-Regulation Mediates Interleukin-24-Induced Apoptosis through Inhibition of Translation. Cancers 2018, 10, 153. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Talukdar, S.; Bhoopathi, P.; Shen, X.-N.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. Mda-7/IL-24 Mediates Cancer Cell-Specific Death via Regulation of miR-221 and the Beclin-1 Axis. Cancer Res. 2017, 77, 949–959. [Google Scholar] [CrossRef]
- Menezes, M.E.; Shen, X.-N.; Das, S.K.; Emdad, L.; Guo, C.; Yuan, F.; Li, Y.-J.; Archer, M.C.; Zacksenhaus, E.; Windle, J.J.; et al. MDA-7/IL-24 Functions as a Tumor Suppressor Gene in Vivo in Transgenic Mouse Models of Breast Cancer. Oncotarget 2015, 6, 36928–36942. [Google Scholar] [CrossRef]
- Ramesh, R.; Ahmed, R.; Munshi, A. Interleukin (IL)-24: Reconfiguring the Tumor Microenvironment for Eliciting Antitumor Response. Adv. Exp. Med. Biol. 2021, 1290, 99–110. [Google Scholar] [CrossRef]
- Panneerselvam, J.; Shanker, M.; Jin, J.; Branch, C.D.; Muralidharan, R.; Zhao, Y.D.; Chada, S.; Munshi, A.; Ramesh, R. Phosphorylation of Interleukin (IL)-24 Is Required for Mediating Its Anti-Cancer Activity. Oncotarget 2015, 6, 16271–16286. [Google Scholar] [CrossRef] [PubMed]
- Sauane, M.; Su, Z.-Z.; Dash, R.; Liu, X.; Norris, J.S.; Sarkar, D.; Lee, S.-G.; Allegood, J.C.; Dent, P.; Spiegel, S.; et al. Ceramide Plays a Prominent Role in MDA-7/IL-24-Induced Cancer-Specific Apoptosis. J. Cell Physiol. 2010, 222, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Happy, M.; Dejoie, J.; Zajac, C.K.; Cortez, B.; Chakraborty, K.; Aderemi, J.; Sauane, M. Sigma 1 Receptor Antagonist Potentiates the Anti-Cancer Effect of P53 by Regulating ER Stress, ROS Production, Bax Levels, and Caspase-3 Activation. Biochem. Biophys. Res. Commun. 2015, 456, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Yacoub, A.; Hamed, H.A.; Park, M.A.; Dash, R.; Bhutia, S.K.; Sarkar, D.; Gupta, P.; Emdad, L.; Lebedeva, I.V.; et al. MDA-7/IL-24 as a Cancer Therapeutic: From Bench to Bedside. Anti-Cancer Drugs 2010, 21, 725–731. [Google Scholar] [CrossRef]
- Yacoub, A.; Mitchell, C.; Hong, Y.; Gopalkrishnan, R.V.; Su, Z.-Z.; Gupta, P.; Sauane, M.; Lebedeva, I.V.; Curiel, D.T.; Mahasreshti, P.J.; et al. MDA-7 Regulates Cell Growth and Radiosensitivity in Vitro of Primary (Non-Established) Human Glioma Cells. Cancer Biol. Ther. 2004, 3, 739–751. [Google Scholar] [CrossRef]
- Sarkar, D.; Su, Z.-Z.; Lebedeva, I.V.; Sauane, M.; Gopalkrishnan, R.V.; Valerie, K.; Dent, P.; Fisher, P.B. Mda-7 (IL-24) Mediates Selective Apoptosis in Human Melanoma Cells by Inducing the Coordinated Overexpression of the GADD Family of Genes by Means of P38 MAPK. Proc. Natl. Acad. Sci. USA 2002, 99, 10054–10059. [Google Scholar] [CrossRef]
- Lebedeva, I.V.; Sarkar, D.; Su, Z.-Z.; Kitada, S.; Dent, P.; Stein, C.A.; Reed, J.C.; Fisher, P.B. Bcl-2 and Bcl-x(L) Differentially Protect Human Prostate Cancer Cells from Induction of Apoptosis by Melanoma Differentiation Associated Gene-7, Mda-7/IL-24. Oncogene 2003, 22, 8758–8773. [Google Scholar] [CrossRef]
- Park, M.A.; Walker, T.; Martin, A.P.; Allegood, J.; Vozhilla, N.; Emdad, L.; Sarkar, D.; Rahmani, M.; Graf, M.; Yacoub, A.; et al. MDA-7/IL-24-Induced Cell Killing in Malignant Renal Carcinoma Cells Occurs by a Ceramide/CD95/PERK-Dependent Mechanism. Mol. Cancer Ther. 2009, 8, 1280–1291. [Google Scholar] [CrossRef]
- Li, R.; Erdamar, S.; Dai, H.; Sayeeduddin, M.; Frolov, A.; Wheeler, T.M.; Ayala, G.E. Cytoplasmic Accumulation of Glycogen Synthase Kinase-3beta Is Associated with Aggressive Clinicopathological Features in Human Prostate Cancer. Anti-Cancer Res. 2009, 29, 2077–2081. [Google Scholar]
- Darrington, R.S.; Campa, V.M.; Walker, M.M.; Bengoa-Vergniory, N.; Gorrono-Etxebarria, I.; Uysal-Onganer, P.; Kawano, Y.; Waxman, J.; Kypta, R.M. Distinct Expression and Activity of GSK-3α and GSK-3β in Prostate Cancer. Int. J. Cancer 2012, 131, E872–E883. [Google Scholar] [CrossRef] [PubMed]
- Domoto, T.; Uehara, M.; Bolidong, D.; Minamoto, T. Glycogen Synthase Kinase 3β in Cancer Biology and Treatment. Cells 2020, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Pollak, M.; Topisirovic, I. The Role of GSK3 in Metabolic Pathway Perturbations in Cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119059. [Google Scholar] [CrossRef] [PubMed]
- Chiara, F.; Rasola, A. GSK-3 and Mitochondria in Cancer Cells. Front. Oncol. 2013, 3, 16. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-kappaB Activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Zou, Q.; Li, S.; Zhang, F. KLF3 Transcription Activates WNT1 and Promotes the Growth and Metastasis of Gastric Cancer via Activation of the WNT/β-Catenin Signaling Pathway. Lab. Investig. 2023, 103, 100078. [Google Scholar] [CrossRef]
- Shah, K.; Kazi, J.U. Phosphorylation-Dependent Regulation of WNT/Beta-Catenin Signaling. Front. Oncol. 2022, 12, 858782. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen Inactivation of Glycogen Synthase Kinase-3 Beta in Intact Cells via Serine 9 Phosphorylation. Biochem. J. 1994, 303 Pt 3, 701–704. [Google Scholar] [CrossRef]
- Cohen, P.; Goedert, M. GSK3 Inhibitors: Development and Therapeutic Potential. Nat. Rev. Drug Discov. 2004, 3, 479–487. [Google Scholar] [CrossRef]
- Tan, J.; Zhuang, L.; Leong, H.-S.; Iyer, N.G.; Liu, E.T.; Yu, Q. Pharmacologic Modulation of Glycogen Synthase Kinase-3beta Promotes P53-Dependent Apoptosis through a Direct Bax-Mediated Mitochondrial Pathway in Colorectal Cancer Cells. Cancer Res. 2005, 65, 9012–9020. [Google Scholar] [CrossRef]
- Thapa, R.; Gupta, G.; Bhat, A.A.; Almalki, W.H.; Alzarea, S.I.; Kazmi, I.; Saleem, S.; Khan, R.; Altwaijry, N.; Dureja, H.; et al. A Review of Glycogen Synthase Kinase-3 (GSK3) Inhibitors for Cancers Therapies. Int. J. Biol. Macromol. 2023, 253, 127375. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Enomoto, A.; Yoshizaki-Ogawa, A.; Sato, S.; Miyagawa, K.; Yoshizaki, A. The Role of Mammalian STK38 in DNA Damage Response and Targeting for Radio-Sensitization. Cancers 2023, 15, 2054. [Google Scholar] [CrossRef] [PubMed]
- Sokolosky, M.; Chappell, W.H.; Stadelman, K.; Abrams, S.L.; Davis, N.M.; Steelman, L.S.; McCubrey, J.A. Inhibition of GSK-3β Activity Can Result in Drug and Hormonal Resistance and Alter Sensitivity to Targeted Therapy in MCF-7 Breast Cancer Cells. Cell Cycle 2014, 13, 820–833. [Google Scholar] [CrossRef]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by P38 MAPK as an Alternative Pathway for GSK3beta Inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef]
- Valiyari, S.; Salami, M.; Mahdian, R.; Shokrgozar, M.A.; Oloomi, M.; Mohammadi Farsani, A.; Bouzari, S. sIL-24 Peptide, a Human Interleukin-24 Isoform, Induces Mitochondrial-Mediated Apoptosis in Human Cancer Cells. Cancer Chemother. Pharmacol. 2017, 80, 451–459. [Google Scholar] [CrossRef]
- Kroon, J.; in ’t Veld, L.S.; Buijs, J.T.; Cheung, H.; van der Horst, G.; van der Pluijm, G. Glycogen Synthase Kinase-3β Inhibition Depletes the Population of Prostate Cancer Stem/Progenitor-like Cells and Attenuates Metastatic Growth. Oncotarget 2014, 5, 8986–8994. [Google Scholar] [CrossRef]
- Liao, X.; Thrasher, J.B.; Holzbeierlein, J.; Stanley, S.; Li, B. Glycogen Synthase Kinase-3beta Activity Is Required for Androgen-Stimulated Gene Expression in Prostate Cancer. Endocrinology 2004, 145, 2941–2949. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, J.; Han, S.; Liu, J.; Holzbeierlein, J.; Thrasher, J.B.; Li, B. Suppression of Glycogen Synthase Kinase 3 Activity Reduces Tumor Growth of Prostate Cancer in Vivo. Prostate 2011, 71, 835–845. [Google Scholar] [CrossRef]
- Haga, N.; Saito, S.; Tsukumo, Y.; Sakurai, J.; Furuno, A.; Tsuruo, T.; Tomida, A. Mitochondria Regulate the Unfolded Protein Response Leading to Cancer Cell Survival under Glucose Deprivation Conditions. Cancer Sci. 2010, 101, 1125–1132. [Google Scholar] [CrossRef]
- Ma, L.; Wei, J.; Wan, J.; Wang, W.; Wang, L.; Yuan, Y.; Yang, Z.; Liu, X.; Ming, L. Low Glucose and Metformin-Induced Apoptosis of Human Ovarian Cancer Cells Is Connected to ASK1 via Mitochondrial and Endoplasmic Reticulum Stress-Associated Pathways. J. Exp. Clin. Cancer Res. 2019, 38, 77. [Google Scholar] [CrossRef]
- Harrington, C.T.; Sotillo, E.; Robert, A.; Hayer, K.E.; Bogusz, A.M.; Psathas, J.; Yu, D.; Taylor, D.; Dang, C.V.; Klein, P.S.; et al. Transient Stabilization, Rather than Inhibition, of MYC Amplifies Extrinsic Apoptosis and Therapeutic Responses in Refractory B-Cell Lymphoma. Leukemia 2019, 33, 2429–2441. [Google Scholar] [CrossRef] [PubMed]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J.; et al. Glycogen Synthase Kinase-3 Inhibition Induces Glioma Cell Death through c-MYC, Nuclear Factor-kappaB, and Glucose Regulation. Cancer Res. 2008, 68, 6643–6651. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, A.; Lopez, S.; Smith, S.; Sony, A.; Abreu, J.; de la Parra, C.; Sauane, M. Interleukin 24 Promotes Mitochondrial Dysfunction, Glucose Regulation, and Apoptosis by Inactivating Glycogen Synthase Kinase 3 Beta in Human Prostate Cancer Cells. Cells 2025, 14, 357. https://doi.org/10.3390/cells14050357
Kim A, Lopez S, Smith S, Sony A, Abreu J, de la Parra C, Sauane M. Interleukin 24 Promotes Mitochondrial Dysfunction, Glucose Regulation, and Apoptosis by Inactivating Glycogen Synthase Kinase 3 Beta in Human Prostate Cancer Cells. Cells. 2025; 14(5):357. https://doi.org/10.3390/cells14050357
Chicago/Turabian StyleKim, Anastassiya, Sual Lopez, Simira Smith, Alphons Sony, Jennifer Abreu, Columba de la Parra, and Moira Sauane. 2025. "Interleukin 24 Promotes Mitochondrial Dysfunction, Glucose Regulation, and Apoptosis by Inactivating Glycogen Synthase Kinase 3 Beta in Human Prostate Cancer Cells" Cells 14, no. 5: 357. https://doi.org/10.3390/cells14050357
APA StyleKim, A., Lopez, S., Smith, S., Sony, A., Abreu, J., de la Parra, C., & Sauane, M. (2025). Interleukin 24 Promotes Mitochondrial Dysfunction, Glucose Regulation, and Apoptosis by Inactivating Glycogen Synthase Kinase 3 Beta in Human Prostate Cancer Cells. Cells, 14(5), 357. https://doi.org/10.3390/cells14050357