
Role of Epigenetics in Chronic Lung Disease

 , and
, and {kind=link}
{kind=link}
Abstract
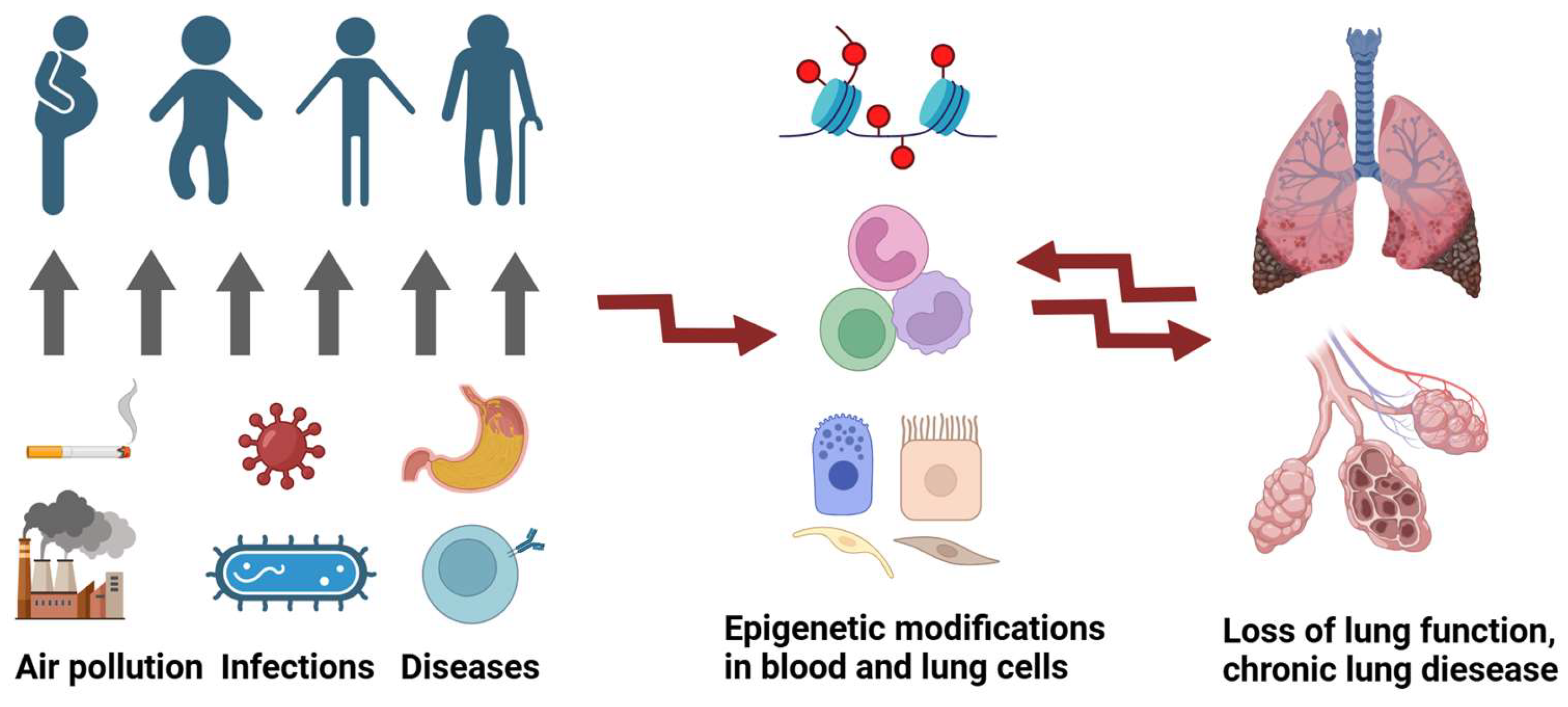
1. Introduction
2. COPD
3. Asthma
4. IPF
5. Cystic Fibrosis
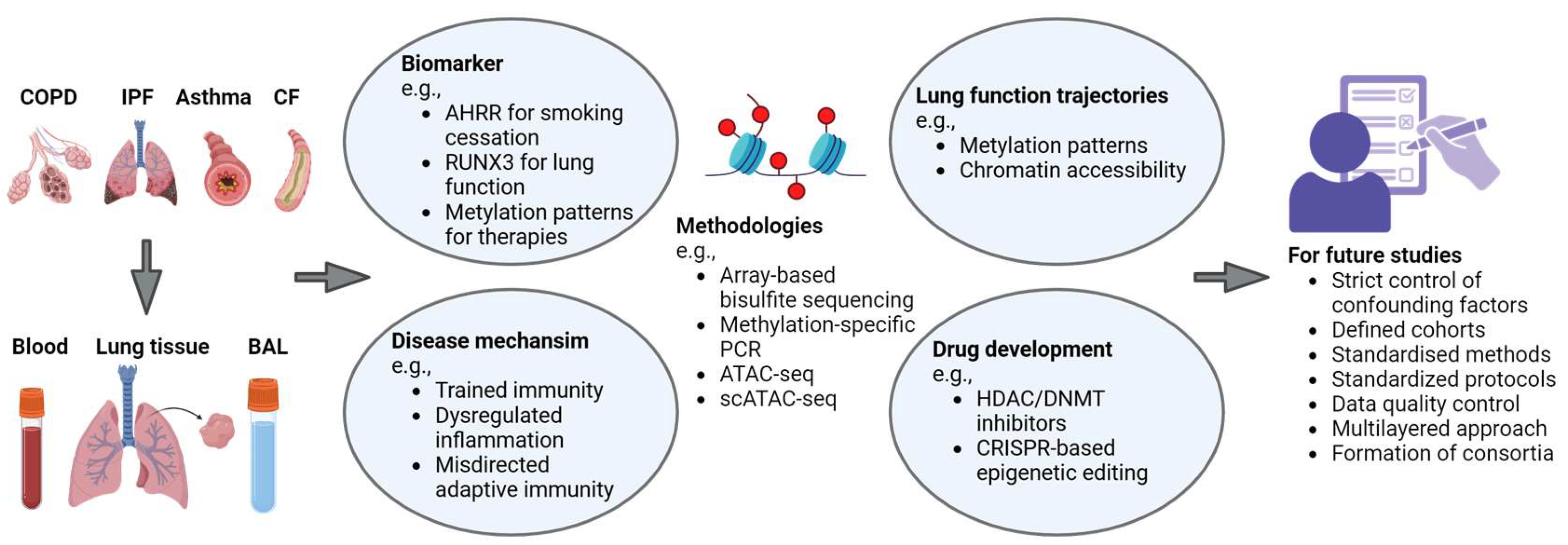
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ritzmann, F.; Lunding, L.P.; Bals, R.; Wegmann, M.; Beisswenger, C. IL-17 Cytokines and Chronic Lung Diseases. Cells 2022, 11, 2132. [Google Scholar] [CrossRef]
- Rabe, K.F.; Hurd, S.; Anzueto, A.; Barnes, P.J.; Buist, S.A.; Calverley, P.; Fukuchi, Y.; Jenkins, C.; Rodriguez-Roisin, R.; van Weel, C.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2007, 176, 532–555. [Google Scholar] [CrossRef]
- Tsukui, T.; Wolters, P.J.; Sheppard, D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature 2024, 631, 627–634. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Dupont, S.; Wickstrom, S.A. Mechanical regulation of chromatin and transcription. Nat. Rev. Genet. 2022, 23, 624–643. [Google Scholar] [CrossRef]
- Ermolaeva, M.; Neri, F.; Ori, A.; Rudolph, K.L. Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 594–610. [Google Scholar] [CrossRef]
- Agusti, A.; Melen, E.; DeMeo, D.L.; Breyer-Kohansal, R.; Faner, R. Pathogenesis of chronic obstructive pulmonary disease: Understanding the contributions of gene-environment interactions across the lifespan. Lancet Respir. Med. 2022, 10, 512–524. [Google Scholar] [CrossRef]
- Casas-Recasens, S.; Cassim, R.; Mendoza, N.; Agusti, A.; Lodge, C.; Li, S.; Bui, D.; Martino, D.; Dharmage, S.C.; Faner, R. Epigenome-Wide Association Studies of COPD and Lung Function: A Systematic Review. Am. J. Respir. Crit. Care Med. 2024, 210, 766–778. [Google Scholar] [CrossRef]
- Kachroo, P.; Morrow, J.D.; Kho, A.T.; Vyhlidal, C.A.; Silverman, E.K.; Weiss, S.T.; Tantisira, K.G.; DeMeo, D.L. Co-methylation analysis in lung tissue identifies pathways for fetal origins of COPD. Eur. Respir. J. 2020, 56, 1902347. [Google Scholar] [CrossRef]
- Martino, D.J.; Bui, D.S.; Li, S.; Idrose, S.; Perret, J.; Lowe, A.J.; Lodge, C.J.; Bowatte, G.; Moodley, Y.; Thomas, P.S.; et al. Genetic and Epigenetic Associations with Pre-Chronic Obstructive Pulmonary Disease Lung Function Trajectories. Am. J. Respir. Crit. Care Med. 2023, 208, 1135–1137. [Google Scholar] [CrossRef]
- Kaur, G.; Begum, R.; Thota, S.; Batra, S. A systematic review of smoking-related epigenetic alterations. Arch. Toxicol. 2019, 93, 2715–2740. [Google Scholar] [CrossRef]
- Morrow, J.D.; Make, B.; Regan, E.; Han, M.; Hersh, C.P.; Tal-Singer, R.; Quackenbush, J.; Choi, A.M.K.; Silverman, E.K.; DeMeo, D.L. DNA Methylation Is Predictive of Mortality in Current and Former Smokers. Am. J. Respir. Crit. Care Med. 2020, 201, 1099–1109. [Google Scholar] [CrossRef]
- Tsaprouni, L.G.; Yang, T.P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Vinuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Lee, M.K.; Hong, Y.; Kim, S.Y.; London, S.J.; Kim, W.J. DNA methylation and smoking in Korean adults: Epigenome-wide association study. Clin. Epigenetics 2016, 8, 103. [Google Scholar] [CrossRef]
- Mao, Y.; Huang, P.; Wang, Y.; Wang, M.; Li, M.D.; Yang, Z. Genome-wide methylation and expression analyses reveal the epigenetic landscape of immune-related diseases for tobacco smoking. Clin. Epigenetics 2021, 13, 215. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S.; et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef]
- Saint-Andre, V.; Charbit, B.; Biton, A.; Rouilly, V.; Posseme, C.; Bertrand, A.; Rotival, M.; Bergstedt, J.; Patin, E.; Albert, M.L.; et al. Smoking changes adaptive immunity with persistent effects. Nature 2024, 626, 827–835. [Google Scholar] [CrossRef]
- Guida, F.; Sandanger, T.M.; Castagne, R.; Campanella, G.; Polidoro, S.; Palli, D.; Krogh, V.; Tumino, R.; Sacerdote, C.; Panico, S.; et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 2015, 24, 2349–2359. [Google Scholar] [CrossRef]
- Ambatipudi, S.; Cuenin, C.; Hernandez-Vargas, H.; Ghantous, A.; Le Calvez-Kelm, F.; Kaaks, R.; Barrdahl, M.; Boeing, H.; Aleksandrova, K.; Trichopoulou, A.; et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics 2016, 8, 599–618. [Google Scholar] [CrossRef]
- Skov-Jeppesen, S.M.; Kobylecki, C.J.; Jacobsen, K.K.; Bojesen, S.E. Changing Smoking Behavior and Epigenetics: A Longitudinal Study of 4,432 Individuals from the General Population. Chest 2023, 163, 1565–1575. [Google Scholar] [CrossRef]
- Imboden, M.; Wielscher, M.; Rezwan, F.I.; Amaral, A.F.S.; Schaffner, E.; Jeong, A.; Beckmeyer-Borowko, A.; Harris, S.E.; Starr, J.M.; Deary, I.J.; et al. Epigenome-wide association study of lung function level and its change. Eur. Respir. J. 2019, 54, 1900457. [Google Scholar] [CrossRef]
- Carmona, J.J.; Barfield, R.T.; Panni, T.; Nwanaji-Enwerem, J.C.; Just, A.C.; Hutchinson, J.N.; Colicino, E.; Karrasch, S.; Wahl, S.; Kunze, S.; et al. Metastable DNA methylation sites associated with longitudinal lung function decline and aging in humans: An epigenome-wide study in the NAS and KORA cohorts. Epigenetics 2018, 13, 1039–1055. [Google Scholar] [CrossRef]
- Philibert, R.; Hollenbeck, N.; Andersen, E.; McElroy, S.; Wilson, S.; Vercande, K.; Beach, S.R.; Osborn, T.; Gerrard, M.; Gibbons, F.X.; et al. Reversion of AHRR Demethylation Is a Quantitative Biomarker of Smoking Cessation. Front. Psychiatry 2016, 7, 55. [Google Scholar] [CrossRef]
- Wilson, R.; Wahl, S.; Pfeiffer, L.; Ward-Caviness, C.K.; Kunze, S.; Kretschmer, A.; Reischl, E.; Peters, A.; Gieger, C.; Waldenberger, M. The dynamics of smoking-related disturbed methylation: A two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genom. 2017, 18, 805. [Google Scholar] [CrossRef]
- Hillary, R.F.; McCartney, D.L.; Smith, H.M.; Bernabeu, E.; Gadd, D.A.; Chybowska, A.D.; Cheng, Y.; Murphy, L.; Wrobel, N.; Campbell, A.; et al. Blood-based epigenome-wide analyses of 19 common disease states: A longitudinal, population-based linked cohort study of 18,413 Scottish individuals. PLoS Med. 2023, 20, e1004247. [Google Scholar] [CrossRef]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef]
- Bermingham, M.L.; Walker, R.M.; Marioni, R.E.; Morris, S.W.; Rawlik, K.; Zeng, Y.; Campbell, A.; Redmond, P.; Whalley, H.C.; Adams, M.J.; et al. Identification of novel differentially methylated sites with potential as clinical predictors of impaired respiratory function and COPD. EBioMedicine 2019, 43, 576–586. [Google Scholar] [CrossRef]
- Lee, M.; Huan, T.; McCartney, D.L.; Chittoor, G.; de Vries, M.; Lahousse, L.; Nguyen, J.N.; Brody, J.A.; Castillo-Fernandez, J.; Terzikhan, N.; et al. Pulmonary Function and Blood DNA Methylation: A Multiancestry Epigenome-Wide Association Meta-analysis. Am. J. Respir. Crit. Care Med. 2022, 206, 321–336. [Google Scholar] [CrossRef]
- de Vries, M.; van der Plaat, D.A.; Vonk, J.M.; Boezen, H.M. No association between DNA methylation and COPD in never and current smokers. BMJ Open Respir. Res. 2018, 5, e000282. [Google Scholar] [CrossRef]
- Buro-Auriemma, L.J.; Salit, J.; Hackett, N.R.; Walters, M.S.; Strulovici-Barel, Y.; Staudt, M.R.; Fuller, J.; Mahmoud, M.; Stevenson, C.S.; Hilton, H.; et al. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum. Mol. Genet. 2013, 22, 4726–4738. [Google Scholar] [CrossRef]
- Vucic, E.A.; Chari, R.; Thu, K.L.; Wilson, I.M.; Cotton, A.M.; Kennett, J.Y.; Zhang, M.; Lonergan, K.M.; Steiling, K.; Brown, C.J.; et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am. J. Respir. Cell Mol. Biol. 2014, 50, 912–922. [Google Scholar] [CrossRef]
- Hernandez Cordero, A.I.; Yang, C.X.; Yang, J.; Horvath, S.; Shaipanich, T.; MacIsaac, J.; Lin, D.T.S.; Kobor, M.S.; Guillemi, S.; Harris, M.; et al. Airway Aging and Methylation Disruptions in HIV-associated Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2022, 206, 150–160. [Google Scholar] [CrossRef]
- Morrow, J.D.; Cho, M.H.; Hersh, C.P.; Pinto-Plata, V.; Celli, B.; Marchetti, N.; Criner, G.; Bueno, R.; Washko, G.; Glass, K.; et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics 2016, 11, 730–739. [Google Scholar] [CrossRef]
- Sundar, I.K.; Yin, Q.; Baier, B.S.; Yan, L.; Mazur, W.; Li, D.; Susiarjo, M.; Rahman, I. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin. Epigenetics 2017, 9, 38. [Google Scholar] [CrossRef]
- Casas-Recasens, S.; Noell, G.; Mendoza, N.; Lopez-Giraldo, A.; Garcia, T.; Guirao, A.; Agusti, A.; Faner, R. Lung DNA Methylation in Chronic Obstructive Pulmonary Disease: Relationship with Smoking Status and Airflow Limitation Severity. Am. J. Respir. Crit. Care Med. 2021, 203, 129–134. [Google Scholar] [CrossRef]
- Ringh, M.V.; Hagemann-Jensen, M.; Needhamsen, M.; Kular, L.; Breeze, C.E.; Sjoholm, L.K.; Slavec, L.; Kullberg, S.; Wahlstrom, J.; Grunewald, J.; et al. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine 2019, 46, 290–304. [Google Scholar] [CrossRef]
- Eriksson Strom, J.; Kebede Merid, S.; Pourazar, J.; Blomberg, A.; Lindberg, A.; Ringh, M.V.; Hagemann-Jensen, M.; Ekstrom, T.J.; Behndig, A.F.; Melen, E. Chronic Obstructive Pulmonary Disease Is Associated with Epigenome-Wide Differential Methylation in BAL Lung Cells. Am. J. Respir. Cell Mol. Biol. 2022, 66, 638–647. [Google Scholar] [CrossRef]
- Groth, E.E.; Weber, M.; Bahmer, T.; Pedersen, F.; Kirsten, A.; Bornigen, D.; Rabe, K.F.; Watz, H.; Ammerpohl, O.; Goldmann, T. Exploration of the sputum methylome and omics deconvolution by quadratic programming in molecular profiling of asthma and COPD: The road to sputum omics 2.0. Respir. Res. 2020, 21, 274. [Google Scholar] [CrossRef]
- Clifford, R.L.; Fishbane, N.; Patel, J.; MacIsaac, J.L.; McEwen, L.M.; Fisher, A.J.; Brandsma, C.A.; Nair, P.; Kobor, M.S.; Hackett, T.L.; et al. Altered DNA methylation is associated with aberrant gene expression in parenchymal but not airway fibroblasts isolated from individuals with COPD. Clin. Epigenetics 2018, 10, 32. [Google Scholar] [CrossRef]
- Schwartz, U.; Llamazares Prada, M.; Pohl, S.T.; Richter, M.; Tamas, R.; Schuler, M.; Keller, C.; Mijosek, V.; Muley, T.; Schneider, M.A.; et al. High-resolution transcriptomic and epigenetic profiling identifies novel regulators of COPD. EMBO J. 2023, 42, e111272. [Google Scholar] [CrossRef]
- Hernandez Cordero, A.I.; Yang, C.X.; Milne, S.; Li, X.; Hollander, Z.; Chen, V.; Ng, R.; Tebbutt, S.J.; Leung, J.M.; Sin, D.D. Epigenetic blood biomarkers of ageing and mortality in COPD. Eur. Respir. J. 2021, 58, 2101890. [Google Scholar] [CrossRef]
- Hillary, R.F.; Stevenson, A.J.; McCartney, D.L.; Campbell, A.; Walker, R.M.; Howard, D.M.; Ritchie, C.W.; Horvath, S.; Hayward, C.; McIntosh, A.M.; et al. Epigenetic measures of ageing predict the prevalence and incidence of leading causes of death and disease burden. Clin. Epigenetics 2020, 12, 115. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Levanen, B.; Bhakta, N.R.; Torregrosa Paredes, P.; Barbeau, R.; Hiltbrunner, S.; Pollack, J.L.; Skold, C.M.; Svartengren, M.; Grunewald, J.; Gabrielsson, S.; et al. Altered microRNA profiles in bronchoalveolar lavage fluid exosomes in asthmatic patients. J. Allergy Clin. Immunol. 2013, 131, 894–903. [Google Scholar] [CrossRef]
- Kho, A.T.; Sharma, S.; Davis, J.S.; Spina, J.; Howard, D.; McEnroy, K.; Moore, K.; Sylvia, J.; Qiu, W.; Weiss, S.T.; et al. Circulating MicroRNAs: Association with Lung Function in Asthma. PLoS ONE 2016, 11, e0157998. [Google Scholar] [CrossRef]
- Davis, J.S.; Sun, M.; Kho, A.T.; Moore, K.G.; Sylvia, J.M.; Weiss, S.T.; Lu, Q.; Tantisira, K.G. Circulating microRNAs and association with methacholine PC20 in the Childhood Asthma Management Program (CAMP) cohort. PLoS ONE 2017, 12, e0180329. [Google Scholar] [CrossRef]
- Seumois, G.; Chavez, L.; Gerasimova, A.; Lienhard, M.; Omran, N.; Kalinke, L.; Vedanayagam, M.; Ganesan, A.P.; Chawla, A.; Djukanovic, R.; et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat. Immunol. 2014, 15, 777–788. [Google Scholar] [CrossRef]
- Weiss, S.T.; Van Natta, M.L.; Zeiger, R.S. Relationship between increased airway responsiveness and asthma severity in the childhood asthma management program. Am. J. Respir. Crit. Care Med. 2000, 162, 50–56. [Google Scholar] [CrossRef]
- Van Asselt, A.J.; Beck, J.J.; Finnicum, C.T.; Johnson, B.N.; Kallsen, N.; Viet, S.; Huizenga, P.; Ligthart, L.; Hottenga, J.J.; Pool, R.; et al. Epigenetic signatures of asthma: A comprehensive study of DNA methylation and clinical markers. Clin. Epigenetics 2024, 16, 151. [Google Scholar] [CrossRef]
- Arathimos, R.; Suderman, M.; Sharp, G.C.; Burrows, K.; Granell, R.; Tilling, K.; Gaunt, T.R.; Henderson, J.; Ring, S.; Richmond, R.C.; et al. Epigenome-wide association study of asthma and wheeze in childhood and adolescence. Clin. Epigenetics 2017, 9, 112. [Google Scholar] [CrossRef]
- Yang, I.V.; Pedersen, B.S.; Liu, A.; O’Connor, G.T.; Teach, S.J.; Kattan, M.; Misiak, R.T.; Gruchalla, R.; Steinbach, S.F.; Szefler, S.J.; et al. DNA methylation and childhood asthma in the inner city. J. Allergy Clin. Immunol. 2015, 136, 69–80. [Google Scholar] [CrossRef]
- Nicodemus-Johnson, J.; Naughton, K.A.; Sudi, J.; Hogarth, K.; Naurekas, E.T.; Nicolae, D.L.; Sperling, A.I.; Solway, J.; White, S.R.; Ober, C. Genome-Wide Methylation Study Identifies an IL-13-induced Epigenetic Signature in Asthmatic Airways. Am. J. Respir. Crit. Care Med. 2016, 193, 376–385. [Google Scholar] [CrossRef]
- Kumar, M.; Ahmad, T.; Sharma, A.; Mabalirajan, U.; Kulshreshtha, A.; Agrawal, A.; Ghosh, B. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 2011, 128, 1077–1085. [Google Scholar] [CrossRef]
- Malmhall, C.; Alawieh, S.; Lu, Y.; Sjostrand, M.; Bossios, A.; Eldh, M.; Radinger, M. MicroRNA-155 is essential for T(H)2-mediated allergen-induced eosinophilic inflammation in the lung. J. Allergy Clin. Immunol. 2014, 133, 1429–1438. [Google Scholar] [CrossRef]
- Stein, M.M.; Hrusch, C.L.; Gozdz, J.; Igartua, C.; Pivniouk, V.; Murray, S.E.; Ledford, J.G.; Marques Dos Santos, M.; Anderson, R.L.; Metwali, N.; et al. Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N. Engl. J. Med. 2016, 375, 411–421. [Google Scholar] [CrossRef]
- DeVries, A.; McCauley, K.; Fadrosh, D.; Fujimura, K.E.; Stern, D.A.; Lynch, S.V.; Vercelli, D. Maternal prenatal immunity, neonatal trained immunity, and early airway microbiota shape childhood asthma development. Allergy 2022, 77, 3617–3628. [Google Scholar] [CrossRef]
- Breton, C.V.; Byun, H.M.; Wenten, M.; Pan, F.; Yang, A.; Gilliland, F.D. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467. [Google Scholar] [CrossRef]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef]
- Perera, F.; Tang, W.Y.; Herbstman, J.; Tang, D.; Levin, L.; Miller, R.; Ho, S.M. Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS ONE 2009, 4, e4488. [Google Scholar] [CrossRef]
- Nieto, A.; Mazon, A.; Nieto, M.; Calderon, R.; Calaforra, S.; Selva, B.; Uixera, S.; Palao, M.J.; Brandi, P.; Conejero, L.; et al. Bacterial Mucosal Immunotherapy with MV130 Prevents Recurrent Wheezing in Children: A Randomized, Double-Blind, Placebo-controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 462–472. [Google Scholar] [CrossRef]
- Jartti, T.; Gern, J.E. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 2017, 140, 895–906. [Google Scholar] [CrossRef]
- Machiels, B.; Dourcy, M.; Xiao, X.; Javaux, J.; Mesnil, C.; Sabatel, C.; Desmecht, D.; Lallemand, F.; Martinive, P.; Hammad, H.; et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat. Immunol. 2017, 18, 1310–1320. [Google Scholar] [CrossRef]
- Chen, P.C.; Shao, Y.T.; Hsieh, M.H.; Kao, H.F.; Kuo, W.S.; Wang, S.M.; Chen, S.H.; Wu, L.S.H.; Tsai, H.J.; Wang, J.Y. Early-life EV-A71 infection augments allergen-induced airway inflammation in asthma through trained macrophage immunity. Cell Mol. Immunol. 2021, 18, 472–483. [Google Scholar] [CrossRef]
- Rajput, C.; Han, M.; Ishikawa, T.; Lei, J.; Jazaeri, S.; Bentley, J.K.; Hershenson, M.B. Early-life heterologous rhinovirus infections induce an exaggerated asthma-like phenotype. J. Allergy Clin. Immunol. 2020, 146, 571–582. [Google Scholar] [CrossRef]
- Steer, C.A.; Matha, L.; Shim, H.; Takei, F. Lung group 2 innate lymphoid cells are trained by endogenous IL-33 in the neonatal period. JCI Insight 2020, 5, e135961. [Google Scholar] [CrossRef]
- Verma, M.; Verma, D.; Sripada, A.S.; Sirohi, K.; Varma, R.; Sahu, A.; Alam, R. NFkappaB1 inhibits memory formation and supports effector function of ILC2s in memory-driven asthma. Front. Immunol. 2023, 14, 1217776. [Google Scholar] [CrossRef]
- Eljaszewicz, A.; Ruchti, F.; Radzikowska, U.; Globinska, A.; Boonpiyathad, T.; Gschwend, A.; Morita, H.; Helbling, A.; Arasi, S.; Kahlert, H.; et al. Trained immunity and tolerance in innate lymphoid cells, monocytes, and dendritic cells during allergen-specific immunotherapy. J. Allergy Clin. Immunol. 2021, 147, 1865–1877. [Google Scholar] [CrossRef]
- Lechner, A.; Henkel, F.D.R.; Hartung, F.; Bohnacker, S.; Alessandrini, F.; Gubernatorova, E.O.; Drutskaya, M.S.; Angioni, C.; Schreiber, Y.; Haimerl, P.; et al. Macrophages acquire a TNF-dependent inflammatory memory in allergic asthma. J. Allergy Clin. Immunol. 2022, 149, 2078–2090. [Google Scholar] [CrossRef]
- Han, X.; Liu, L.; Huang, S.; Xiao, W.; Gao, Y.; Zhou, W.; Zhang, C.; Zheng, H.; Yang, L.; Xie, X.; et al. RNA m(6)A methylation modulates airway inflammation in allergic asthma via PTX3-dependent macrophage homeostasis. Nat. Commun. 2023, 14, 7328. [Google Scholar] [CrossRef]
- Stefanowicz, D.; Hackett, T.L.; Garmaroudi, F.S.; Gunther, O.P.; Neumann, S.; Sutanto, E.N.; Ling, K.M.; Kobor, M.S.; Kicic, A.; Stick, S.M.; et al. DNA methylation profiles of airway epithelial cells and PBMCs from healthy, atopic and asthmatic children. PLoS ONE 2012, 7, e44213. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, S.W.; Kim, T.H.; Park, J.S.; Cheong, H.S.; Shin, H.D.; Park, C.S. Genome-wide methylation profiling of the bronchial mucosa of asthmatics: Relationship to atopy. BMC Med. Genet. 2013, 14, 39. [Google Scholar] [CrossRef]
- Stefanowicz, D.; Lee, J.Y.; Lee, K.; Shaheen, F.; Koo, H.K.; Booth, S.; Knight, D.A.; Hackett, T.L. Elevated H3K18 acetylation in airway epithelial cells of asthmatic subjects. Respir. Res. 2015, 16, 95. [Google Scholar] [CrossRef]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More Than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Dwyer, D.F.; Nyquist, S.K.; Buchheit, K.M.; Vukovic, M.; Deb, C.; Wadsworth, M.H., 2nd; Hughes, T.K.; Kazer, S.W.; Yoshimoto, E.; et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 2018, 560, 649–654. [Google Scholar] [CrossRef]
- Bigot, J.; Guillot, L.; Guitard, J.; Ruffin, M.; Corvol, H.; Chignard, M.; Hennequin, C.; Balloy, V. Respiratory Epithelial Cells Can Remember Infection: A Proof-of-Concept Study. J. Infect. Dis. 2020, 221, 1000–1005. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed]
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic pulmonary fibrosis beyond the lung: Understanding disease mechanisms to improve diagnosis and management. Respir. Res. 2021, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef]
- Lee, J.U.; Son, J.H.; Shim, E.Y.; Cheong, H.S.; Shin, S.W.; Shin, H.D.; Baek, A.R.; Ryu, S.; Park, C.S.; Chang, H.S.; et al. Global DNA Methylation Pattern of Fibroblasts in Idiopathic Pulmonary Fibrosis. DNA Cell Biol. 2019, 38, 905–914. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef]
- Hanmandlu, A.; Zhu, L.; Mertens, T.C.J.; Collum, S.; Bi, W.; Xiong, F.; Wang, R.; Amirthalingam, R.T.; Ren, D.; Han, L.; et al. Transcriptomic and Epigenetic Profiling of Fibroblasts in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 53–63. [Google Scholar] [CrossRef]
- McErlean, P.; Bell, C.G.; Hewitt, R.J.; Busharat, Z.; Ogger, P.P.; Ghai, P.; Albers, G.J.; Calamita, E.; Kingston, S.; Molyneaux, P.L.; et al. DNA Methylome Alterations Are Associated with Airway Macrophage Differentiation and Phenotype during Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 954–966. [Google Scholar] [CrossRef]
- Nakano, Y.; Yang, I.V.; Walts, A.D.; Watson, A.M.; Helling, B.A.; Fletcher, A.A.; Lara, A.R.; Schwarz, M.I.; Evans, C.M.; Schwartz, D.A. MUC5B Promoter Variant rs35705950 Affects MUC5B Expression in the Distal Airways in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 464–466. [Google Scholar] [CrossRef]
- Borie, R.; Cardwell, J.; Konigsberg, I.R.; Moore, C.M.; Zhang, W.; Sasse, S.K.; Gally, F.; Dobrinskikh, E.; Walts, A.; Powers, J.; et al. Colocalization of Gene Expression and DNA Methylation with Genetic Risk Variants Supports Functional Roles of MUC5B and DSP in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 1259–1270. [Google Scholar] [CrossRef]
- Gally, F.; Sasse, S.K.; Kurche, J.S.; Gruca, M.A.; Cardwell, J.H.; Okamoto, T.; Chu, H.W.; Hou, X.; Poirion, O.B.; Buchanan, J.; et al. The MUC5B-associated variant rs35705950 resides within an enhancer subject to lineage- and disease-dependent epigenetic remodeling. JCI Insight 2021, 6, e144294. [Google Scholar] [CrossRef]
- Valenzi, E.; Bahudhanapati, H.; Tan, J.; Tabib, T.; Sullivan, D.I.; Nouraie, M.; Sembrat, J.; Fan, L.; Chen, K.; Liu, S.; et al. Single-nucleus chromatin accessibility identifies a critical role for TWIST1 in idiopathic pulmonary fibrosis myofibroblast activity. Eur. Respir. J. 2023, 62, 2200474. [Google Scholar] [CrossRef]
- Velagacherla, V.; Mehta, C.H.; Nayak, Y.; Nayak, U.Y. Molecular pathways and role of epigenetics in the idiopathic pulmonary fibrosis. Life Sci. 2022, 291, 120283. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Chen, Y.; Armstrong, D.A.; Salas, L.A.; Hazlett, H.F.; Nymon, A.B.; Dessaint, J.A.; Aridgides, D.S.; Mellinger, D.L.; Liu, X.; Christensen, B.C.; et al. Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin. Epigenetics 2018, 10, 152. [Google Scholar] [CrossRef]
- Magalhaes, M.; Tost, J.; Pineau, F.; Rivals, I.; Busato, F.; Alary, N.; Mely, L.; Leroy, S.; Murris, M.; Caimmi, D.; et al. Dynamic changes of DNA methylation and lung disease in cystic fibrosis: Lessons from a monogenic disease. Epigenomics 2018, 10, 1131–1145. [Google Scholar] [CrossRef]
- Scott, M.; De Sario, A. DNA methylation changes in cystic fibrosis: Cause or consequence? Clin. Genet. 2020, 98, 3–9. [Google Scholar] [CrossRef]
- Magalhaes, M.; Rivals, I.; Claustres, M.; Varilh, J.; Thomasset, M.; Bergougnoux, A.; Mely, L.; Leroy, S.; Corvol, H.; Guillot, L.; et al. DNA methylation at modifier genes of lung disease severity is altered in cystic fibrosis. Clin. Epigenetics 2017, 9, 19. [Google Scholar] [CrossRef]
- Kabadi, A.M.; Machlin, L.; Dalal, N.; Lee, R.E.; McDowell, I.; Shah, N.N.; Drowley, L.; Randell, S.H.; Reddy, T.E. Epigenome editing of the CFTR-locus for treatment of cystic fibrosis. J. Cyst. Fibros. 2022, 21, 164–171. [Google Scholar] [CrossRef]
- Caution, K.; Pan, A.; Krause, K.; Badr, A.; Hamilton, K.; Vaidya, A.; Gosu, H.; Daily, K.; Estfanous, S.; Gavrilin, M.A.; et al. Methylomic correlates of autophagy activity in cystic fibrosis. J. Cyst. Fibros. 2019, 18, 491–500. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritzmann, F.; Brand, M.; Bals, R.; Wegmann, M.; Beisswenger, C. Role of Epigenetics in Chronic Lung Disease. Cells 2025, 14, 251. https://doi.org/10.3390/cells14040251
Ritzmann F, Brand M, Bals R, Wegmann M, Beisswenger C. Role of Epigenetics in Chronic Lung Disease. Cells. 2025; 14(4):251. https://doi.org/10.3390/cells14040251
Chicago/Turabian StyleRitzmann, Felix, Michelle Brand, Robert Bals, Michael Wegmann, and Christoph Beisswenger. 2025. "Role of Epigenetics in Chronic Lung Disease" Cells 14, no. 4: 251. https://doi.org/10.3390/cells14040251
APA StyleRitzmann, F., Brand, M., Bals, R., Wegmann, M., & Beisswenger, C. (2025). Role of Epigenetics in Chronic Lung Disease. Cells, 14(4), 251. https://doi.org/10.3390/cells14040251