Abstract
The mechanism of kidney injury associated with apolipoprotein L1 (APOL1) risk variants has remained elusive. Complicating this issue is the broad clinical spectrum of APOL1 kidney disease, which has engendered speculation that this reflects multiple mechanisms of kidney injury. APOL1 kidney disease can be rapid in onset with heavy proteinuria, associated with viral infections and categorized pathologically as collapsing focal segmental glomerulosclerosis. Alternatively, APOL1 kidney disease also may present as an insidious, slowly progressive disease, with less proteinuria but losses in glomerular filtration rate and with varied pathology. In addition to APOL1 kidney disease, APOL1 risk variants are also associated with preeclampsia and other conditions related to placental insufficiency. The outcome of these APOL1-associated pregnancy complications frequently results in prematurity and low birth weight, both of which are known risk factors for hypertension and kidney disease later in life due to reduced nephron endowment. The significance of APOL1 risk variants on pregnancy complications that predispose to kidney disease should not be overlooked as a central mechanism of APOL1 kidney disease, especially the insidious forms, which are difficult to distinguish from the spectrum of kidney disease attributable to prematurity and low birth weight. If low nephron endowment is a causal mechanism behind some forms of APOL1 kidney disease, this may have an impact on clinical trials evaluating drugs directly inhibiting APOL1, since in these instances, ongoing APOL1 expression may not be driving podocyte loss and progressive kidney dysfunction.
1. Introduction
A breakthrough in 2010 identified the genetic basis for the enhanced risk for chronic kidney disease (CKD) associated with African ancestry [1,2,3]. Polymorphisms in the gene for apolipoprotein L1 (APOL1, gene name: APOL1) constituting two allelic variants known as G1 and G2 were associated with CKD when inherited as a recessive trait. These risk variants were enriched in individuals of recent sub-Saharan West African ancestry due to their beneficial protection against lethal parasitic infections but are absent in other demographic groups. Over the past 15 years, how APOL1 risk variants cause injury to the kidney and how best to treat individuals with APOL1 kidney disease have been areas of intense research efforts. An early experimental attempt to develop a small animal model for human APOL1 expression serendipitously discovered that APOL1 risk alleles are also expressed in the placenta and cause preeclampsia [4], another disease with excess risk in mothers of African descent [5,6,7]. Several human cohort studies have now confirmed APOL1 high-risk genotypes in the infant predispose the mother to preeclampsia, resulting in infant prematurity and low birth weight [8,9,10,11,12,13]. The potential for this pregnancy complication to have a significant impact on the presentation of APOL1 kidney disease and a comparison to the current disease paradigm of direct effects of APOL1 on kidney cells are the focus of this review.
2. APOL1 and CKD Risk
The current theory is APOL1 kidney disease pathogenesis is a two-hit mechanism [14]. The first hit, the APOL1 high-risk genotype, consists of any combination of two copies of the G1 or G2 variant alleles. The high-risk genotype, however, is only a genetic susceptibility and alone is insufficient to cause CKD. Individuals with high-risk genotypes remain disease-free until a second hit stressor is encountered. The CKDs most strongly associated with APOL1 risk variants are triggered by viral infections, such as human immunodeficiency virus-associated nephropathy (HIVAN) [15,16,17,18,19], coronavirus disease-associated nephropathy (COVAN) [20,21,22,23,24], and others [25]. Mechanistically, immune responses to the viral infection, which involve secretion of high levels of interferons and other pro-inflammatory cytokines, induce APOL1 expression in podocytes [26]. This high level of APOL1 expression subsequently causes podocyte loss by either cell death or detachment leading to the prototypical pathology seen in APOL1 kidney disease: focal segmental glomerulosclerosis (FSGS). Exactly how APOL1 biochemically engages cellular pathways to cause podocyte loss is not fully understood, and numerous causal pathways have been implicated [27,28,29], in addition to potential complicating issues relating to splice variants, cell types, and haplotype effects [30,31,32]. This lack of a unified understanding of the function of APOL1 in kidney cells, in combination with a limited understanding of the potential range of disease-causing second hits, has fueled speculation that the varied presentation of APOL1 kidney diseases is attributable to a complex interaction of genetic, cellular, and environmental factors [30].
There are, however, some common points of understanding in the penetrance and phenotype variability in the presentation of APOL1 kidney disease [30,33]. First, the two-hit model described above is based on APOL1 kidney diseases in which the second hit stressor is known, those caused by viral infections. These APOL1 kidney diseases are overt and relatively rapid in onset, with nephrotic range proteinuria and with the severe FSGS pathology of glomerular collapse (Table 1). These infection-associated presentations have the highest odds ratios for CKD risk in cohort studies (reviewed in [33]). In cases of HIV-associated nephropathy, suppressing the viral infection with antiretroviral therapy, thereby eliminating the second hit from the pathogenic equation, can restore kidney function and pathology [34]. Details on the mechanism of the viral second hit were further revealed in experiments [35,36,37] and human observational reports [38]. The common theme is that the anti-viral immune response, not the virus itself, is likely the critical factor for disease induction since administration of interferons alone can similarly cause kidney disease. However, it also has been shown in model systems using interferon stimulation to induce APOL1 expression that kidney injury can be mitigated using agents to directly suppress APOL1 [37,39]. Therefore, kidney injury appears to be caused by a combination of APOL1 risk-variant expression in the setting of a strong interferon response. It is important to note pre-clinical experiments supporting the development of therapeutics to treat APOL1 kidney disease, including those currently in clinical trials, were based on this two-hit, interferon-dependent model [37,39,40,41].
The second common point of understanding in the phenotype variability is that most presentations of APOL1 kidney disease do not have an obvious second hit [2,42,43,44,45,46,47,48]. These CKDs are insidious, with unclear disease onset, characterized functionally by a subclinical decline in estimated glomerular filtration rate (eGFR) but without severe proteinuria and with a more varied pathological picture (Table 1). The insidious presentations have lower odds ratios in cohort studies compared to the viral-induced overt presentations. This failure to understand the more common, insidious presentation of APOL1 kidney disease has resulted in speculation on various potential mechanisms. One mechanism previously proposed by several groups expands on the two-hit interferon model [49,50]. In this potential scenario, instead of a single strong and sustained interferon-producing event, there are multiple, but less severe and transient events, such as infection with common cold or flu viruses, resulting in short-term and limited episodes of podocyte losses. Thus, in individuals with APOL1 high-risk genotypes, the life-long accumulation of typical anti-viral responses could amass excessive podocyte losses with age. This would then result in a more gradual functional decline but still with progression to a clinical CKD diagnosis faster or more frequently than individuals with low-risk genotypes. There is no direct evidence for this proposed mechanism, but future studies using available mouse models may be helpful to test this hypothesis. In addition, all forms of CKD can be impacted by common co-morbidities, such as hypertension, diabetes or metabolic abnormalities, and environmental and lifestyle factors, such as smoking and alcohol use, which may synergize to hasten functional declines by mechanisms independent of APOL1 expression. However, an alternative potential explanation of the insidious presentation may lie in the association of APOL1 risk alleles with pregnancy complications.

Table 1.
Comparison of the CKD presentations for APOL1 kidney disease with CKDs associated with prematurity and low birth weight.
Table 1.
Comparison of the CKD presentations for APOL1 kidney disease with CKDs associated with prematurity and low birth weight.
| Overt Presentation | Insidious Presentation | CKD from Prematurity or Low Birth Weight | |
|---|---|---|---|
| CKD onset | severe/rapid | subclinical/chronic | subclinical/chronic |
| Clinical presentation | nephrotic range proteinuria | eGFR decline | eGFR decline |
| eGFR decline | hypertension | hypertension | |
| proteinuria | microalbuminuria | ||
| Pathology | FSGS/collapsing FSGS | Nephrosclerosis, microcysts | Nephrosclerosis, |
| Podocytopathy [18,20,51] | FSGS, IFTA [44,52,53] | FSGS, IFTA [54,55] | |
| Diagnoses | HIVAN [15,16,17,18] | HTN-attributed CKD or ESKD [2,42,43,44,45] | HTN-attributed CKD [56] |
| COVAN [20,21,22,23,24] | non-diabetic ESKD [46,47,48] | glomerular disease/nephritis interstitial nephritis [57,58] | |
| Interferon therapy use [38] | primary CKD progression (LN, MN, SCN) [59,60,61,62,63,64] | primary CKD progression (IgAN, PKD, MN, MCD, DN) [65,66,67,68] | |
| FSGS [2,16,43,69] | secondary to AKI [70,71] | ||
| CKD odds ratio | 17–89 (African ancestry) [33] | 2–11 (African ancestry) [33] | 1.2–6 (all races) [57,72,73,74,75,76] |
AKI, acute kidney injury; CKD, chronic kidney disease; COVAN, coronavirus disease-associated nephropathy; DN, diabetic nephropathy; ESKD, end stage kidney disease; FSGS, focal segmental glomerulosclerosis; HIVAN, human immunodeficiency virus associated nephropathy; HTN, hypertension; IFTA, interstitial fibrosis tubular atrophy; IgAN, IgA nephropathy; LN, lupus nephritis; MCD, minimal change disease; MN, membranous nephropathy; PKD, polycystic kidney disease; SCN, sickle cell nephropathy.
3. APOL1 and Risk for Preeclampsia, Prematurity, and Low Birth Weight
As noted above, animal models of human APOL1 expression revealed the potential contribution of APOL1 risk variants to preeclampsia [4,77]. These studies compared mice transgenic for the non-disease APOL1 G0 with the disease-associated G1 or G2 alleles, including one mouse model that replicated the endogenous expression of APOL1 [77]. Since mice do not have a gene equivalent to human APOL1, the transgenic mice expressing either the G1 or G2 allele are a genocopy for humans homozygous for either G1 or G2, and thus these studies essentially examined the effects of APOL1 high-risk genotypes. Both studies documented typical features of preeclampsia in the mice expressing either the G1 or G2 alleles but not in the G0 transgenic mice, including elevated blood pressure and profound outcomes on the morbidity and mortality of both the pregnant female (eclamptic seizures) and in the offspring pups, including perinatal mortality and low birth weight. An interesting caveat of these observations was that the risk of preeclampsia was dependent on the APOL1 genotype of the fetus not the mother, implicating an important contribution of the paternal APOL1 genotype to preeclampsia risk. APOL1 is abundantly expressed in the placenta [78], with high levels of expression in trophoblasts [4], key cell types required for the development of the placental vasculature and maintenance of the maternal–fetal blood barrier [79]. Preeclampsia is caused by the failure of spiral artery remodeling to low resistance/high flow blood vessels resulting in under perfusion of the placenta. This leads to placental insufficiency, a common pathogenic event in various disorders that can result in fetal growth restriction and low birth weight, still birth, and prematurity. The function of APOL1 in the placenta and the mechanism by which APOL1 risk variants lead to placental insufficiency and pregnancy complications are unknown. In speculation, a possible mechanism for APOL1-associated preeclampsia may be similar to the APOL1 risk variant cytotoxicity described for podocyte cell death in CKD [14]. Trophoblast migration into the myometrium and invasion of maternal spiral arteries is required to initiate their vascular remodeling to adequately supply blood flow to the placenta and support the growing fetus [80]. If APOL1 risk variant expression similarly results in trophoblast cell death, there may be too few trophoblasts to induce spiral artery remodeling, setting in motion the well-known mechanism for preeclampsia and placental insufficiency [80].
Similar to CKD, the incidence of prematurity and low birth weight in African Americans is significantly greater than that of European Americans (Table 2). Current United States national vital statistic birth data report African Americans have a greater incidence of prematurity (<37 weeks), low birth weight (<2500 g), and preeclampsia [81]. Similar numbers also apply to other populations of Black African ancestry, with reports of elevated risk of preeclampsia or low birth weight in Caribbean Hispanics and in south and west African countries [82,83,84,85]. Cohort studies of African Americans, other African diasporas, and sub-Saharan African communities found fetal APOL1 high-risk genotype predisposes the mother to risk for preeclampsia and infants born with low birth weight [8,9,10,11,12,13]. The risk for preeclampsia, prematurity, or low birth weight was specific to the fetal APOL1 genotype and did not associate with the maternal APOL1 genotype [10,11,12,86]. When excluding other psychosocial and economic confounders that can contribute to pregnancy complications, the fetal APOL1 high-risk genotype still accounts for a significant portion of the excess risk of preeclampsia in African ancestry compared to European ancestry [10]. Although preeclamptic pregnancies frequently result in preterm birth, in a large cohort of term preeclamptic pregnancies, infants with the high-risk APOL1 genotype showed altered fetal growth velocity and had significantly higher risk of being small for gestational age (<10th percentile in birth weight) [9]. This suggests that in less severe cases of preeclampsia and independent of prematurity, these pregnancies are characterized by an adverse intrauterine environment secondary to placental insufficiency. These observations together implicate the fetal APOL1 risk genotype as a significant risk factor for both prematurity and low birth weight.
Table 2.
Impact of APOL1 high-risk genotype on incidence and risk of preeclampsia and low birth weight in the United States.
4. Pregnancy Complications, Nephron Endowment, and CKD Risk
Prematurity and low birth weight are well-established risk factors for CKD, decreased eGFR, and hypertension in both children and adults [57,72,74,76,91,92]. The CKDs associated with prematurity and low birth weight have similar features to the insidious presentation of APOL1 kidney disease (Table 1). These CKDs can have varied diagnoses but frequently there is no identifiable cause [56]. They can become evident in childhood or adulthood, typically with unclear disease onset, but they are progressive with a common disease pathology of glomerulonephritis or FSGS [57,58]. Odds ratios for CKD risk range similarly to the insidious presentations of APOL1 kidney disease. Connections between APOL1 genotype, kidney disease, and prematurity or low birth weight have been examined by multiple groups using the NEPTUNE, CKiD, and CureGN consortia [93,94,95]. Prematurity was significantly more common in children and adults with APOL1 high-risk genotypes, typically with a glomerular disease diagnosis, lower eGFR at study entry, and faster eGFR decline compared to an APOL1 low-risk genotype.
The mechanism for CKD caused by prematurity and low birth weight originates in kidney development (reviewed in [96]). Nephron endowment is determined at birth and establishes the maximum functional capacity of the kidneys. Since a majority of nephrogenesis takes place in the third trimester, prematurity can significantly interrupt kidney development and reduce total nephron numbers. Low birth weight and smallforgestationalage status are determined by both the length of gestation and the quality of the intrauterine environment. Therefore, in addition to prematurity, an adverse intrauterine environment can both reduce birth weight and kidney size or maturation [97].
Brenner originally proposed that low nephron endowment can be the origin of kidney disease and hypertension in later life [98]. This work arose from observations on the classic experimental model known as five-sixths nephrectomy, in which after partial nephrectomy, the remnant kidney is subject to maladaptive responses [99]. To compensate for low nephron numbers, the existing nephrons hyperfilter and hypertrophy, reducing sodium excretion and altering glomerular hemodynamics. These adaptations lessen the ability of the kidney to withstand stress (i.e., functional kidney reserve), the outcome of which is hypertension and risk for CKD. Experimental rodent models controlling nephron number or podocyte loss demonstrated podocyte attrition was a central component to nephron functional decline [54,100,101]. Since glomerular podocytes are terminally differentiated and do not undergo mitosis, the increase in glomerular size requires existing podocytes to stretch over the hypertrophied glomerulus causing mechanical strain on the podocyte. This strain places the podocyte at risk for detachment or cell death, further reducing podocyte density in the glomerulus, which perpetuates continued podocyte loss and causes the typical scar formation of FSGS.
Biopsy and autopsy studies have linked prematurity and low birth weight with low glomeruli numbers, increased glomerular volume, and low podocyte density per glomerulus in subjects with and without a diagnosed CKD [58,102,103]. Recent studies have begun examining APOL1 genotype effects on nephron numbers and podocyte densities. In mouse models of human APOL1 expression (including mouse models replicating native APOL1 expression), APOL1 risk alleles were associated with larger glomerular volumes (indicating hypertrophy) and lower podocyte densities (indicating podocyte loss) compared to mice expressing APOL1 G0 [4,104]. These were the same transgenic mouse models that also exhibited preeclampsia and low birth weights with APOL1 risk allele expression. In human studies, examination of kidneys from healthy transplant donors observed APOL1 high-risk genotype donors had larger glomeruli and fewer podocyte numbers compared to low-risk genotypes [105]. In a larger cohort of African Americans without a clinical CKD diagnosis, APOL1 high-risk genotypes similarly were associated with losses of podocytes and increases in glomerular volumes with age [106]. Neither of the human studies directly linked APOL1 genotype and low nephron endowment with a birth complication, specifically prematurity and low birth weight, and future studies making this connection with birth history are needed. Continued experiments in the existing APOL1 transgenic mice should be useful in testing this mechanism, and newer imaging and morphometric methods have made estimating nephron numbers in humans more feasible [107].
5. Summary
In the varied presentation of APOL1 kidney disease, the two-hit model of pathogenesis best fits the overt CKDs associated with viral infections, whereas the insidious presentation may be explained, in part, by low nephron endowment caused by pregnancy complications (Figure 1). In both potential mechanisms the endpoint of the pathogenic process culminates in loss of podocytes, resulting in similar FSGS pathology. In the overt presentation, podocyte loss is driven by ongoing APOL1 expression in the kidney; however in the insidious presentation, podocyte loss is driven by mechanical strain in the glomerulus. In the insidious presentation, the APOL1-dependent event that caused CKD was expression of APOL1 in the placenta, and thus this mechanism would not require any direct effect of APOL1 expression after pregnancy. It is possible to still consider the low nephron endowment mechanism a two-hit model. Low nephron endowment at birth is frequently considered a susceptibility event since the reduced nephron number limits the functional reserve of the kidney to withstand disease stressors or even normal aging. Thus, low nephron endowment can be a first-hit susceptibility to other more common insults associated with CKD risk, such as hypertension and diabetes. The magnitude, frequency, or compounding of these second-hit CKD risk factors may further accelerate nephron loss and result in variabilities in CKD presentation, severity, or progression.
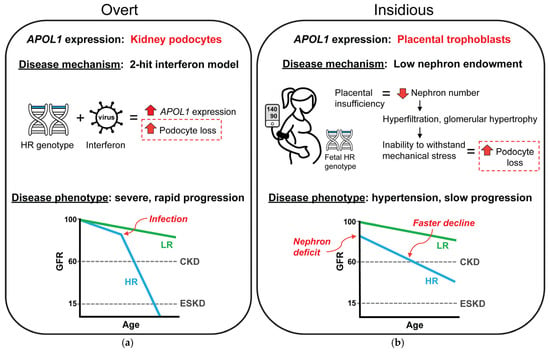
Figure 1.
Proposed mechanistic difference between the varied presentations of APOL1 kidney disease. (a) The best understood presentation of APOL1 kidney disease is an overt, rapidly progressive CKD that is typically associated with a viral infection or other chronic inflammatory state. These inflammatory conditions are associated with high interferon levels which induce APOL1 expression in kidney podocytes resulting in dramatic podocyte losses and nephrotic range proteinuria. The overt presentation best fits the proposed two-hit model for APOL1 kidney disease pathogenesis. (b) Less understood is the insidious presentation of APOL1 kidney disease, a subclinical disease that is characterized by slowly progressive losses in GFR, frequently with hypertension. This presentation of APOL1 kidney disease is similar to CKD that can develop as a consequence of prematurity and low birth weight through a mechanism caused by low nephron endowment. A developmental deficit in nephron numbers predisposes kidneys to gradual, chronic podocyte loss from compensatory mechanisms related to countering the mechanical strain during filtration. Thus, with the evidence for APOL1 expression in placental trophoblasts and the association of fetal APOL1 high-risk genotypes with preeclampsia and low birth weight, it is possible that a similar CKD mechanism for podocyte loss may underlie the insidious presentation of APOL1 kidney disease. In this potential mechanism, podocyte losses would not require ongoing expression of APOL1 in the kidney but are founded in the effect of APOL1 expression during pregnancy and also would not require a second hit to cause CKD. HR, high-risk APOL1 genotype; LR, low-risk APOL1 genotype, CKD, chronic kidney disease, ESKD, end stage kidney disease, GFR, glomerular filtration rate.
If low nephron endowment is a contributor to some presentations of insidious APOL1 kidney disease, the current anti-APOL1 drugs in clinical testing may be ineffective in these cases since ongoing APOL1 expression is not driving podocyte loss and the ensuing proteinuria and eGFR decline. These new anti-APOL1 therapeutics represent a long-needed advance for kidney diseases that have previously lacked any specific therapeutics and disproportionally impact underserved populations [108]. However, if low nephron endowment does contribute to insidious APOL1 kidney disease, then these clinical trials may have some non-responders. Based on the existing studies examining APOL1 risk genotypes and prematurity and low birth weight (summarized in Table 2), approximately 20% of individuals with APOL1 high-risk genotype experienced significant birth complications that could be the underlying cause for their CKD. Establishing a direct cause–effect relationship connecting (1) APOL1 risk genotype with pregnancy complications, (2) pregnancy complications with low nephron endowment, and (3) low nephron endowment with CKD may be challenging and may remain only an association. However, prematurity and low birth weight are well-recognized as significant contributors to CKD risk later in life, and evidence is growing associating APOL1 genotypes with pregnancy complications that can cause low nephron endowment. For future cohort studies and clinical trials, it will be difficult to separate CKDs caused by APOL1-associated birth complications from those caused by ongoing APOL1 expression in the kidney unless information is collected on birth history.
6. Conclusions
The high incidence and significance of pregnancy-related birth complications caused by APOL1 risk genotypes are potentially an under-recognized contributor for CKD risk. The varied presentations of APOL1 kidney disease may in part result from placental APOL1 expression and its effect on nephron endowment, since the insidious presentation of APOL1 kidney disease has many similarities to kidney disease attributed to prematurity and low birth weight. If the mechanism of low nephron endowment underlies insidious APOL1 kidney disease, clinical trials testing anti-APOL1 drugs may not be efficacious in these cases since ongoing APOL1 expression in the kidney would not be driving podocyte injury and loss. As recently discussed, clinical assessment of birth history is underutilized in evaluating CKD risk [109]. If APOL1 genotyping is used to guide management of future CKD risk, an accompanying evaluation of birth history could provide a more comprehensive understanding of the long-term impact of APOL1 genotype and treatment options.
Author Contributions
Conceptualization L.A.B.; writing—original draft preparation, L.A.B. and T.A.; writing—review and editing L.A.B. and T.A.; funding acquisition, L.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
L.A.B. was supported by National Institutes of Health grants, numbers 1R01DK127638-05, 1R01DK135265-04.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created for this manuscript.
Acknowledgments
We thank Katherine Dell and Jeffrey Schelling for critical review of the manuscript.
Conflicts of Interest
L.A.B. has received royalties for APOL1 research tools. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| AKI | acute kidney injury |
| APOL1 | Apolipoprotein L1 |
| COVAN | coronavirus disease-associated nephropathy |
| CKD | chronic kidney disease |
| CKiD | Chronic Kidney Disease in Children Study |
| CureGN | Cure Glomerulonephropathy Consortium |
| DN | diabetic nephropathy |
| eGFR | estimated glomerular filtration rate |
| ESKD | end stage kidney disease |
| FSGS | focal segmental glomerulosclerosis |
| GFR | glomerular filtration rate |
| HIVAN | human immunodeficiency virus-associated nephropathy |
| HTN | hypertension |
| IFTA | interstitial fibrosis tubular atrophy |
| IgAN | IgA nephropathy |
| LN | lupus nephritis |
| MCD | minimal change disease |
| MN | membranous nephropathy |
| NEPTUNE | Nephrotic Syndrome Study Network |
| PKD | polycystic kidney disease |
| SCN | sickle cell nephropathy |
References
- Tzur, S.; Rosset, S.; Shemer, R.; Yudkovsky, G.; Selig, S.; Tarekegn, A.; Bekele, E.; Bradman, N.; Wasser, W.G.; Behar, D.M.; et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum. Genet. 2010, 128, 345–350. [Google Scholar] [CrossRef]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef]
- Genovese, G.; Tonna, S.J.; Knob, A.U.; Appel, G.B.; Katz, A.; Bernhardy, A.J.; Needham, A.W.; Lazarus, R.; Pollak, M.R. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. 2010, 78, 698–704. [Google Scholar] [CrossRef]
- Bruggeman, L.A.; Wu, Z.; Luo, L.; Madhavan, S.M.; Konieczkowski, M.; Drawz, P.E.; Thomas, D.B.; Barisoni, L.; Sedor, J.R.; O’Toole, J.F. APOL1-G0 or APOL1-G2 Transgenic Models Develop Preeclampsia but Not Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 3600–3610. [Google Scholar] [CrossRef]
- Conti-Ramsden, F.; de Marvao, A.; Gill, C.; Chappell, L.C.; Myers, J.; Vuckovic, D.; Dehghan, A.; Hysi, P.G. Association of genetic ancestry with pre-eclampsia in multi-ethnic cohorts of pregnant women. Pregnancy Hypertens. 2024, 38, 101162. [Google Scholar] [CrossRef]
- Croke, L. Gestational Hypertension and Preeclampsia: A Practice Bulletin from ACOG. Am. Fam. Physician 2019, 100, 649–650. [Google Scholar]
- Noubiap, J.J.; Bigna, J.J.; Nyaga, U.F.; Jingi, A.M.; Kaze, A.D.; Nansseu, J.R.; Fokom Domgue, J. The burden of hypertensive disorders of pregnancy in Africa: A systematic review and meta-analysis. J. Clin. Hypertens. 2019, 21, 479–488. [Google Scholar] [CrossRef]
- Miller, A.K.; Azhibekov, T.; O’Toole, J.F.; Sedor, J.R.; Williams, S.M.; Redline, R.W.; Bruggeman, L.A. Association of preeclampsia with infant APOL1 genotype in African Americans. BMC Med. Genet. 2020, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Azhibekov, T.; Durodoye, R.; Miller, A.K.; Simpson, C.L.; Davis, R.L.; Williams, S.M.; Bruggeman, L.A. Fetal High-Risk APOL1 Genotype Increases Risk for Small for Gestational Age in Term Infants Affected by Preeclampsia. Neonatology 2023, 120, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.J.; Hjorten, R.C.; Simpson, C.L.; Rosenberg, A.Z.; Rosenblum, S.D.; Kovesdy, C.P.; Tylavsky, F.A.; Myrie, J.; Ruiz, B.L.; Haque, S.; et al. Fetal-Not Maternal-APOL1 Genotype Associated with Risk for Preeclampsia in Those with African Ancestry. Am. J. Hum. Genet. 2018, 103, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Rosenberg, A.Z.; Zhang, B.; Binns-Roemer, E.; David, V.; Lv, Y.; Hjorten, R.C.; Reidy, K.J.; Chen, T.K.; Wang, G.; et al. Joint Associations of Maternal-Fetal APOL1 Genotypes and Maternal Country of Origin with Preeclampsia Risk. Am. J. Kidney Dis. 2021, 77, 879–888.e871. [Google Scholar] [CrossRef]
- Sheehy, S.; Friedman, D.; Liu, C.; Lunetta, K.L.; Zirpoli, G.; Palmer, J.R. Association between Apolipoprotein L1 genetic variants and risk of preeclampsia and preterm birth among U.S. Black women. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2025, 25, 100365. [Google Scholar] [CrossRef] [PubMed]
- Thakoordeen-Reddy, S.; Winkler, C.; Moodley, J.; David, V.; Binns-Roemer, E.; Ramsuran, V.; Naicker, T. Maternal variants within the apolipoprotein L1 gene are associated with preeclampsia in a South African cohort of African ancestry. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 246, 129–133. [Google Scholar] [CrossRef]
- Friedman, D.J.; Pollak, M.R. APOL1 and Kidney Disease: From Genetics to Biology. Annu. Rev. Physiol. 2020, 82, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Atta, M.G.; Estrella, M.M.; Kuperman, M.; Foy, M.C.; Fine, D.M.; Racusen, L.C.; Lucas, G.M.; Nelson, G.W.; Warner, A.C.; Winkler, C.A.; et al. HIV-associated nephropathy patients with and without apolipoprotein L1 gene variants have similar clinical and pathological characteristics. Kidney Int. 2012, 82, 338–343. [Google Scholar] [CrossRef]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, A.N.; Duarte, R.; Ramsay, M.; Mosiane, P.; Dickens, C.; Dix-Peek, T.; Limou, S.; Sezgin, E.; Nelson, G.W.; Fogo, A.B.; et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. J. Am. Soc. Nephrol. 2015, 26, 2882–2890. [Google Scholar] [CrossRef]
- Fine, D.M.; Wasser, W.G.; Estrella, M.M.; Atta, M.G.; Kuperman, M.; Shemer, R.; Rajasekaran, A.; Tzur, S.; Racusen, L.C.; Skorecki, K. APOL1 risk variants predict histopathology and progression to ESRD in HIV-related kidney disease. J. Am. Soc. Nephrol. 2012, 23, 343–350. [Google Scholar] [CrossRef]
- Kudose, S.; Santoriello, D.; Bomback, A.S.; Stokes, M.B.; Batal, I.; Markowitz, G.S.; Wyatt, C.M.; D’Agati, V.D. The spectrum of kidney biopsy findings in HIV-infected patients in the modern era. Kidney Int. 2020, 97, 1006–1016. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Wu, Z.; Agudelo, J.; Herlitz, L.C.; Miller, A.W.; Bruggeman, L.A. Local Inflammation But Not Kidney Cell Infection Associated with High APOL1 Expression in COVID-Associated Nephropathy. Kidney360 2023, 4, 1757–1762. [Google Scholar] [CrossRef]
- Shetty, A.A.; Tawhari, I.; Safar-Boueri, L.; Seif, N.; Alahmadi, A.; Gargiulo, R.; Aggarwal, V.; Usman, I.; Kisselev, S.; Gharavi, A.G.; et al. COVID-19-Associated Glomerular Disease. J. Am. Soc. Nephrol. 2020, 32, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Larsen, C.P.; Hernandez-Arroyo, C.F.; Mohamed, M.M.B.; Caza, T.; Sharshir, M.; Chughtai, A.; Xie, L.; Gimenez, J.M.; Sandow, T.A.; et al. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL 1 High-Risk Genotype. J. Am. Soc. Nephrol. 2020, 31, 1688–1695. [Google Scholar] [CrossRef]
- May, R.M.; Cassol, C.; Hannoudi, A.; Larsen, C.P.; Lerma, E.V.; Haun, R.S.; Braga, J.R.; Hassen, S.I.; Wilson, J.; VanBeek, C.; et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int. 2021, 100, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Kudose, S.; Batal, I.; Santoriello, D.; Xu, K.; Barasch, J.; Peleg, Y.; Canetta, P.; Ratner, L.E.; Marasa, M.; Gharavi, A.G.; et al. Kidney Biopsy Findings in Patients with COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1959–1968. [Google Scholar] [CrossRef]
- Muehlig, A.K.; Gies, S.; Huber, T.B.; Braun, F. Collapsing Focal Segmental Glomerulosclerosis in Viral Infections. Front. Immunol. 2021, 12, 800074. [Google Scholar] [CrossRef]
- Nichols, B.; Jog, P.; Lee, J.H.; Blackler, D.; Wilmot, M.; D’Agati, V.; Markowitz, G.; Kopp, J.B.; Alper, S.L.; Pollak, M.R.; et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015, 87, 332–342. [Google Scholar] [CrossRef]
- Khalaila, R.; Skorecki, K. Apolipoprotein L1 (APOL1): Consideration of Molecular Evolution, Interaction with APOL3, and Impact of Splice Isoforms Advances Understanding of Cellular and Molecular Mechanisms of Cell Injury. Cells 2025, 14, 1011. [Google Scholar] [CrossRef]
- Friedman, D.J.; Pollak, M.R. In Search of the Mechanism of APOL1 Kidney Disease. J. Am. Soc. Nephrol. 2024, 35, 815–817. [Google Scholar] [CrossRef]
- Pays, E. The Mechanism of Kidney Disease due to APOL1 Risk Variants: Involvement of Two Distinct Processes. J. Am. Soc. Nephrol. 2024, 35, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.R.; Friedman, D.J. APOL1-associated kidney disease: Modulators of the genotype-phenotype relationship. Curr. Opin. Nephrol. Hypertens. 2025, 34, 191–198. [Google Scholar] [CrossRef]
- Höffken, V.; Braun, D.A.; Pavenstädt, H.; Weide, T. A Cell Biologist’s View on APOL1: What We Know and What We Still Need to Address. Cells 2025, 14, 960. [Google Scholar] [CrossRef]
- Pell, J.; Nagata, S.; Menon, M.C. Nonpodocyte Roles of APOL1 Variants: An Evolving Paradigm. Kidney360 2023, 4, e1325–e1331. [Google Scholar] [CrossRef]
- Ojo, A.O.; Adu, D.; Bramham, K.; Freedman, B.I.; Gbadegesin, R.A.; Ilori, T.O.; Jefferson, N.; Olabisi, O.A.; Susztak, K.; Young, B.A.; et al. APOL1 kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2025, in press. [Google Scholar] [CrossRef]
- Winston, J.A.; Bruggeman, L.A.; Ross, M.D.; Jacobson, J.; Ross, L.; D’Agati, V.D.; Klotman, P.E.; Klotman, M.E. Nephropathy and establishment of a renal reservoir of HIV type 1 during primary infection. N. Engl. J. Med. 2001, 344, 1979–1984. [Google Scholar] [CrossRef]
- McCarthy, G.M.; Blasio, A.; Donovan, O.G.; Schaller, L.B.; Bock-Hughes, A.; Magraner, J.M.; Suh, J.H.; Tattersfield, C.F.; Stillman, I.E.; Shah, S.S.; et al. Recessive, gain-of-function toxicity in an APOL1 BAC transgenic mouse model mirrors human APOL1 kidney disease. Dis. Model. Mech. 2021, 14, dmm048952. [Google Scholar] [CrossRef] [PubMed]
- Riella, C.V.; McNulty, M.; Ribas, G.T.; Tattersfield, C.F.; Perez-Gill, C.; Eichinger, F.; Kelly, J.; Chun, J.; Subramanian, B.; Guizelini, D.; et al. ADAR regulates APOL1 via A-to-I RNA editing by inhibition of MDA5 activation in a paradoxical biological circuit. Proc. Nat. Acad. Sci. USA 2022, 119, e2210150119. [Google Scholar] [CrossRef] [PubMed]
- Aghajan, M.; Booten, S.L.; Althage, M.; Hart, C.E.; Ericsson, A.; Maxvall, I.; Ochaba, J.; Menschik-Lundin, A.; Hartleib, J.; Kuntz, S.; et al. Antisense oligonucleotide treatment ameliorates IFN-gamma-induced proteinuria in APOL1-transgenic mice. JCI Insight 2019, 4, e126124. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.S.; Nasr, S.H.; Stokes, M.B.; D’Agati, V.D. Treatment with IFN-α, -β, or -γ is associated with collapsing focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2010, 5, 607–615. [Google Scholar] [CrossRef]
- Egbuna, O.; Zimmerman, B.; Manos, G.; Fortier, A.; Chirieac, M.C.; Dakin, L.A.; Friedman, D.J.; Bramham, K.; Campbell, K.; Knebelmann, B.; et al. Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants. N. Engl. J. Med. 2023, 388, 969–979. [Google Scholar] [CrossRef]
- Nystrom, S.E.; Li, G.; Datta, S.; Soldano, K.L.; Silas, D.; Weins, A.; Hall, G.; Thomas, D.B.; Olabisi, O.A. JAK inhibitor blocks COVID-19 cytokine-induced JAK/STAT/APOL1 signaling in glomerular cells and podocytopathy in human kidney organoids. JCI Insight 2022, 7, e157432. [Google Scholar] [CrossRef]
- Sula Karreci, E.; Jacas, S.; Donovan, O.; Pintye, D.; Wiley, N.; Zsengeller, Z.K.; Schlondorff, J.; Alper, S.L.; Friedman, D.J.; Pollak, M.R. Differing sensitivities to angiotensin converting enzyme inhibition of kidney disease mediated by APOL1 high-risk variants G1 and G2. Kidney Int. 2024, 106, 1072–1085. [Google Scholar] [CrossRef]
- Parsa, A.; Kao, W.H.; Xie, D.; Astor, B.C.; Li, M.; Hsu, C.Y.; Feldman, H.I.; Parekh, R.S.; Kusek, J.W.; Greene, T.H.; et al. APOL1 risk variants, race, and progression of chronic kidney disease. N. Engl. J. Med. 2013, 369, 2183–2196. [Google Scholar] [CrossRef]
- Anyaegbu, E.I.; Shaw, A.S.; Hruska, K.A.; Jain, S. Clinical phenotype of APOL1 nephropathy in young relatives of patients with end-stage renal disease. Pediatr. Nephrol. 2015, 30, 983–989. [Google Scholar] [CrossRef]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef]
- Lipkowitz, M.S.; Freedman, B.I.; Langefeld, C.D.; Comeau, M.E.; Bowden, D.W.; Kao, W.H.; Astor, B.C.; Bottinger, E.P.; Iyengar, S.K.; Klotman, P.E.; et al. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013, 83, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Tzur, S.; Rosset, S.; Skorecki, K.; Wasser, W.G. APOL1 allelic variants are associated with lower age of dialysis initiation and thereby increased dialysis vintage in African and Hispanic Americans with non-diabetic end-stage kidney disease. Nephrol. Dial. Transplant. 2012, 27, 1498–1505. [Google Scholar] [CrossRef]
- Freedman, B.I.; Langefeld, C.D.; Turner, J.; Nunez, M.; High, K.P.; Spainhour, M.; Hicks, P.J.; Bowden, D.W.; Reeves-Daniel, A.M.; Murea, M.; et al. Association of APOL1 variants with mild kidney disease in the first-degree relatives of African American patients with non-diabetic end-stage renal disease. Kidney Int. 2012, 82, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Ulasi, I.I.; Tzur, S.; Wasser, W.G.; Shemer, R.; Kruzel, E.; Feigin, E.; Ijoma, C.K.; Onodugo, O.D.; Okoye, J.U.; Arodiwe, E.B.; et al. High population frequencies of APOL1 risk variants are associated with increased prevalence of non-diabetic chronic kidney disease in the Igbo people from south-eastern Nigeria. Nephron Clin. Pract. 2013, 123, 123–128. [Google Scholar] [CrossRef]
- Bruggeman, L.A.; O’Toole, J.F.; Sedor, J.R. APOL1 polymorphisms and kidney disease: Loss-of-function or gain-of-function? Am. J. Physiol. Renal Physiol. 2019, 316, F1–F8. [Google Scholar] [CrossRef]
- Friedman, D.J. COVID-19 and APOL1: Understanding Disease Mechanisms through Clinical Observation. J. Am. Soc. Nephrol. 2021, 32, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Winkler, C.A.; Zhao, X.; Radeva, M.K.; Gassman, J.J.; D’Agati, V.D.; Nast, C.C.; Wei, C.; Reiser, J.; Guay-Woodford, L.M.; et al. Clinical Features and Histology of Apolipoprotein L1-Associated Nephropathy in the FSGS Clinical Trial. J. Am. Soc. Nephrol. 2015, 26, 1443–1448. [Google Scholar] [CrossRef]
- Larsen, C.P.; Beggs, M.L.; Saeed, M.; Ambruzs, J.M.; Cossey, L.N.; Messias, N.C.; Walker, P.D.; Freedman, B.I. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod. Pathol. 2015, 28, 95–102. [Google Scholar] [CrossRef]
- Zee, J.; McNulty, M.T.; Hodgin, J.B.; Zhdanova, O.; Hingorani, S.; Jefferson, J.A.; Gibson, K.L.; Trachtman, H.; Fornoni, A.; Dell, K.M.; et al. APOL1 genotype-associated morphologic changes among patients with focal segmental glomerulosclerosis. Pediatr. Nephrol. 2021, 36, 2747–2757. [Google Scholar] [CrossRef]
- Good, P.I.; Li, L.; Hurst, H.A.; Serrano Herrera, I.; Xu, K.; Rao, M.; Bateman, D.A.; Al-Awqati, Q.; D’Agati, V.D.; Costantini, F.; et al. Low nephron endowment increases susceptibility to renal stress and chronic kidney disease. JCI Insight 2023, 8, e161316. [Google Scholar] [CrossRef]
- Hodgin, J.B.; Rasoulpour, M.; Markowitz, G.S.; D’Agati, V.D. Very low birth weight is a risk factor for secondary focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2009, 4, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lackland, D.T.; Bendall, H.E.; Osmond, C.; Egan, B.M.; Barker, D.J. Low birth weights contribute to high rates of early-onset chronic renal failure in the Southeastern United States. Arch. Intern. Med. 2000, 160, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Vikse, B.E.; Irgens, L.M.; Leivestad, T.; Hallan, S.; Iversen, B.M. Low birth weight increases risk for end-stage renal disease. J. Am. Soc. Nephrol. 2008, 19, 151–157. [Google Scholar] [CrossRef]
- Ikezumi, Y.; Suzuki, T.; Karasawa, T.; Yamada, T.; Hasegawa, H.; Nishimura, H.; Uchiyama, M. Low birthweight and premature birth are risk factors for podocytopenia and focal segmental glomerulosclerosis. Am. J. Nephrol. 2013, 38, 149–157. [Google Scholar] [CrossRef]
- Larsen, C.P.; Beggs, M.L.; Saeed, M.; Walker, P.D. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J. Am. Soc. Nephrol. 2013, 24, 722–725. [Google Scholar] [CrossRef]
- Freedman, B.I.; Langefeld, C.D.; Andringa, K.K.; Croker, J.A.; Williams, A.H.; Garner, N.E.; Birmingham, D.J.; Hebert, L.A.; Hicks, P.J.; Segal, M.S.; et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014, 66, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.P.; Henderson, C.D.; Anguiano, J.; Aiello, C.P.; Collie, M.M.; Moreno, V.; Hu, Y.; Hogan, S.L.; Falk, R.J. Kidney Disease Progression in Membranous Nephropathy among Black Participants with High-Risk APOL1 Genotype. Clin. J. Am. Soc. Nephrol. 2023, 18, 337–343. [Google Scholar] [CrossRef]
- Elliott, M.D.; Marasa, M.; Cocchi, E.; Vena, N.; Zhang, J.Y.; Khan, A.; Krishna Murthy, S.; Bheda, S.; Milo Rasouly, H.; Povysil, G.; et al. Clinical and Genetic Characteristics of CKD Patients with High-Risk APOL1 Genotypes. J. Am. Soc. Nephrol. 2023, 34, 909–919. [Google Scholar] [CrossRef]
- Ashley-Koch, A.E.; Okocha, E.C.; Garrett, M.E.; Soldano, K.; De Castro, L.M.; Jonassaint, J.C.; Orringer, E.P.; Eckman, J.R.; Telen, M.J. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br. J. Haematol. 2011, 155, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Zhang, X.; Shah, B.; Kanias, T.; Gudehithlu, K.P.; Kittles, R.; Machado, R.F.; Arruda, J.A.; Gladwin, M.T.; Singh, A.K.; et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica 2015, 100, 1275–1284. [Google Scholar] [CrossRef]
- Orskov, B.; Christensen, K.B.; Feldt-Rasmussen, B.; Strandgaard, S. Low birth weight is associated with earlier onset of end-stage renal disease in Danish patients with autosomal dominant polycystic kidney disease. Kidney Int. 2012, 81, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Ruggajo, P.; Svarstad, E.; Leh, S.; Marti, H.P.; Reisæther, A.V.; Vikse, B.E. Low Birth Weight and Risk of Progression to End Stage Renal Disease in IgA Nephropathy—A Retrospective Registry-Based Cohort Study. PLoS ONE 2016, 11, e0153819. [Google Scholar] [CrossRef]
- Duncan, R.C.; Bass, P.S.; Garrett, P.J.; Dathan, J.R. Weight at birth and other factors influencing progression of idiopathic membranous nephropathy. Nephrol. Dial. Transplant. 1994, 9, 875. [Google Scholar] [CrossRef]
- Zidar, N.; Avgustin Cavić, M.; Kenda, R.B.; Ferluga, D. Unfavorable course of minimal change nephrotic syndrome in children with intrauterine growth retardation. Kidney Int. 1998, 54, 1320–1323. [Google Scholar] [CrossRef][Green Version]
- Kallash, M.; Wang, Y.; Smith, A.; Trachtman, H.; Gbadegesin, R.; Nester, C.; Canetta, P.; Wang, C.; Hunley, T.E.; Sperati, C.J.; et al. Rapid Progression of Focal Segmental Glomerulosclerosis in Patients with High-Risk APOL1 Genotypes. Clin. J. Am. Soc. Nephrol. 2023, 18, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Selewski, D.T.; Charlton, J.R.; Jetton, J.G.; Guillet, R.; Mhanna, M.J.; Askenazi, D.J.; Kent, A.L. Neonatal Acute Kidney Injury. Pediatrics 2015, 136, e463–e473. [Google Scholar] [CrossRef]
- Starr, M.C.; Hingorani, S.R. Prematurity and future kidney health: The growing risk of chronic kidney disease. Curr. Opin. Pediatr. 2018, 30, 228–235. [Google Scholar] [CrossRef]
- Brathwaite, K.E.; Levy, R.V.; Sarathy, H.; Agalliu, I.; Johns, T.S.; Reidy, K.J.; Fadrowski, J.J.; Schwartz, G.J.; Kaskel, F.J.; Melamed, M.L. Reduced kidney function and hypertension in adolescents with low birth weight, NHANES 1999–2016. Pediatr. Nephrol. 2023, 38, 3071–3082. [Google Scholar] [CrossRef]
- Fan, Z.J.; Lackland, D.T.; Lipsitz, S.R.; Nicholas, J.S. The association of low birthweight and chronic renal failure among Medicaid young adults with diabetes and/or hypertension. Public Health Rep. 2006, 121, 239–244. [Google Scholar] [CrossRef]
- Gjerde, A.; Reisæter, A.V.; Skrunes, R.; Marti, H.P.; Vikse, B.E. Intrauterine Growth Restriction and Risk of Diverse Forms of Kidney Disease during the First 50 Years of Life. Clin. J. Am. Soc. Nephrol. 2020, 15, 1413–1423. [Google Scholar] [CrossRef]
- Ruggajo, P.; Skrunes, R.; Svarstad, E.; Skjærven, R.; Reisæther, A.V.; Vikse, B.E. Familial Factors, Low Birth Weight, and Development of ESRD: A Nationwide Registry Study. Am. J. Kidney Dis. 2016, 67, 601–608. [Google Scholar] [CrossRef]
- Crump, C.; Sundquist, J.; Winkleby, M.A.; Sundquist, K. Preterm birth and risk of chronic kidney disease from childhood into mid-adulthood: National cohort study. Br. Med. J. 2019, 365, l1346. [Google Scholar] [CrossRef]
- Yoshida, T.; Latt, K.Z.; Shrivastav, S.; Lu, H.; Reidy, K.J.; Charlton, J.R.; Zhao, Y.; Winkler, C.A.; Reznik, S.E.; Rosenberg, A.Z.; et al. Preeclampsia in mice carrying fetuses with APOL1 risk variants. bioRxiv 2024. [Google Scholar] [CrossRef]
- Duchateau, P.N.; Pullinger, C.R.; Cho, M.H.; Eng, C.; Kane, J.P. Apolipoprotein L gene family: Tissue-specific expression, splicing, promoter regions; discovery of a new gene. J. Lipid Res. 2001, 42, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Investig. 2004, 114, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: Relationship to clinical outcome. Hypertension 2013, 62, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Osterman, M.J.K.; Hamilton, B.E.; Martin, J.A.; Driscoll, A.K.; Valenzuela, C.P. Births: Final Data for 2022. Natl. Vital. Stat. Rep. 2024, 73, 1–56. [Google Scholar]
- Riella, C.; Siemens, T.A.; Wang, M.; Campos, R.P.; Moraes, T.P.; Riella, L.V.; Friedman, D.J.; Riella, M.C.; Pollak, M.R. APOL1-Associated Kidney Disease in Brazil. Kidney Int. Rep. 2019, 4, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Nakimuli, A.; Chazara, O.; Byamugisha, J.; Elliott, A.M.; Kaleebu, P.; Mirembe, F.; Moffett, A. Pregnancy, parturition and preeclampsia in women of African ancestry. Am. J. Obstet. Gynecol. 2014, 210, 510–520.e511. [Google Scholar] [CrossRef]
- Ray, J.G.; Wanigaratne, S.; Park, A.L.; Bartsch, E.; Dzakpasu, S.; Urquia, M.L. Preterm preeclampsia in relation to country of birth. J. Perinatol. 2016, 36, 718–722. [Google Scholar] [CrossRef]
- Urquia, M.L.; Ying, I.; Glazier, R.H.; Berger, H.; De Souza, L.R.; Ray, J.G. Serious preeclampsia among different immigrant groups. J. Obstet. Gynaecol. Can. 2012, 34, 348–352. [Google Scholar] [CrossRef]
- Robertson, C.C.; Gillies, C.E.; Putler, R.K.B.; Ng, D.; Reidy, K.J.; Crawford, B.; Sampson, M.G. An investigation of APOL1 risk genotypes and preterm birth in African American population cohorts. Nephrol. Dial. Transplant. 2017, 32, 2051–2058. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reynolds, S.A.; Roberts, J.M.; Bodnar, L.M.; Haggerty, C.L.; Youk, A.O.; Catov, J.M. Fetal sex and race modify the predictors of fetal growth. Matern. Child Health J. 2015, 19, 798–810. [Google Scholar] [CrossRef]
- Paré, E.; Parry, S.; McElrath, T.F.; Pucci, D.; Newton, A.; Lim, K.H. Clinical risk factors for preeclampsia in the 21st century. Obstet. Gynecol. 2014, 124, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Jaamaa, G.; Kaiser, M.; Hills, E.; Soim, A.; Zhu, M.; Shcherbatykh, I.Y.; Samelson, R.; Bell, E.; Zdeb, M.; et al. Racial disparity in hypertensive disorders of pregnancy in New York State: A 10-year longitudinal population-based study. Am. J. Public Health 2007, 97, 163–170. [Google Scholar] [CrossRef]
- Zhang, S.; Cardarelli, K.; Shim, R.; Ye, J.; Booker, K.L.; Rust, G. Racial disparities in economic and clinical outcomes of pregnancy among Medicaid recipients. Matern. Child Health J. 2013, 17, 1518–1525. [Google Scholar] [CrossRef]
- Luyckx, V.A.; Perico, N.; Somaschini, M.; Manfellotto, D.; Valensise, H.; Cetin, I.; Simeoni, U.; Allegaert, K.; Vikse, B.E.; Steegers, E.A.; et al. A developmental approach to the prevention of hypertension and kidney disease: A report from the Low Birth Weight and Nephron Number Working Group. Lancet 2017, 390, 424–428. [Google Scholar] [CrossRef]
- Khalsa, D.D.; Beydoun, H.A.; Carmody, J.B. Prevalence of chronic kidney disease risk factors among low birth weight adolescents. Pediatr. Nephrol. 2016, 31, 1509–1516. [Google Scholar] [CrossRef]
- Ng, D.K.; Robertson, C.C.; Woroniecki, R.P.; Limou, S.; Gillies, C.E.; Reidy, K.J.; Winkler, C.A.; Hingorani, S.; Gibson, K.L.; Hjorten, R.; et al. APOL1-associated glomerular disease among African-American children: A collaboration of the Chronic Kidney Disease in Children (CKiD) and Nephrotic Syndrome Study Network (NEPTUNE) cohorts. Nephrol. Dial. Transplant. 2017, 32, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Isaac, J.S.; Troost, J.P.; Wang, Y.; Garrity, K.; Kaskel, F.; Gbadegesin, R.; Reidy, K.J. Association of Preterm Birth with Adverse Glomerular Disease Outcomes in Children and Adults. Clin. J. Am. Soc. Nephrol. 2024, 19, 1016–1024. [Google Scholar] [CrossRef]
- Hingorani, S.; Gibson, K.L.; Xie, Y.; Wang, Y.; Eddy, S.; Hartman, J.; Sampson, M.; Cassol, C.; Thomas, D.; Gipson, D.S.; et al. The association of low birthweight and prematurity on outcomes in children and adults with nephrotic syndrome-a NEPTUNE cohort study. Pediatr. Nephrol. 2023, 38, 3297–3308. [Google Scholar] [CrossRef]
- Sutherland, M.R.; Black, M.J. The impact of intrauterine growth restriction and prematurity on nephron endowment. Nat. Rev. Nephrol. 2023, 19, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Hinchliffe, S.A.; Lynch, M.R.; Sargent, P.H.; Howard, C.V.; Van Velzen, D. The effect of intrauterine growth retardation on the development of renal nephrons. Br. J. Obstet. Gynaecol. 1992, 99, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Chertow, G.M. Congenital oligonephropathy: An inborn cause of adult hypertension and progressive renal injury? Curr. Opin. Nephrol. Hypertens. 1993, 2, 691–695. [Google Scholar]
- Hostetter, T.H.; Olson, J.L.; Rennke, H.G.; Venkatachalam, M.A.; Brenner, B.M. Hyperfiltration in remnant nephrons: A potentially adverse response to renal ablation. Am. J. Physiol. 1981, 241, F85–F93. [Google Scholar] [CrossRef]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef]
- Wiggins, J.E.; Goyal, M.; Sanden, S.K.; Wharram, B.L.; Shedden, K.A.; Misek, D.E.; Kuick, R.D.; Wiggins, R.C. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: Prevention by calorie restriction. J. Am. Soc. Nephrol. 2005, 16, 2953–2966. [Google Scholar] [CrossRef]
- Koike, K.; Ikezumi, Y.; Tsuboi, N.; Kanzaki, G.; Haruhara, K.; Okabayashi, Y.; Sasaki, T.; Ogura, M.; Saitoh, A.; Yokoo, T. Glomerular Density and Volume in Renal Biopsy Specimens of Children with Proteinuria Relative to Preterm Birth and Gestational Age. Clin. J. Am. Soc. Nephrol. 2017, 12, 585–590. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.; Gao, Q.; Liu, G.; Zheng, J.; Ding, F. Preterm birth leads to a decreased number of differentiated podocytes and accelerated podocyte differentiation. Front. Cell Dev. Biol. 2023, 11, 1142929. [Google Scholar] [CrossRef]
- Bruggeman, L.A.; Wu, Z.; Luo, L.; Madhavan, S.; Drawz, P.E.; Thomas, D.B.; Barisoni, L.; O’Toole, J.F.; Sedor, J.R. APOL1-G0 protects podocytes in a mouse model of HIV-associated nephropathy. PLoS ONE 2019, 14, e0224408. [Google Scholar] [CrossRef]
- Chen, D.P.; Zaky, Z.S.; Schold, J.D.; Herlitz, L.C.; El-Rifai, R.; Drawz, P.E.; Bruggeman, L.A.; Barisoni, L.; Hogan, S.L.; Hu, Y.; et al. Podocyte density is reduced in kidney allografts with high-risk APOL1 genotypes at transplantation. Clin. Transplant. 2021, 35, e14234. [Google Scholar] [CrossRef]
- Hoy, W.E.; Hughson, M.D.; Kopp, J.B.; Mott, S.A.; Bertram, J.F.; Winkler, C.A. APOL1 Risk Alleles Are Associated with Exaggerated Age-Related Changes in Glomerular Number and Volume in African-American Adults: An Autopsy Study. J. Am. Soc. Nephrol. 2015, 26, 3179–3189. [Google Scholar] [CrossRef]
- Morozov, D.; Parvin, N.; Conaway, M.; Oxley, G.; Baldelomar, E.J.; Cwiek, A.; deRonde, K.; Beeman, S.C.; Charlton, J.R.; Bennett, K.M. Estimating Nephron Number from Biopsies: Impact on Clinical Studies. J. Am. Soc. Nephrol. 2022, 33, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Sedor, J.R. APOL1 Kidney Disease: Discovery to Targeted Therapy in 10 Years. Clin. J. Am. Soc. Nephrol. 2024, 19, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.B.; Orozco, A.; Shemies, R.; Attini, R.; Cabiddu, G.; Toreggiani, M.; Jesudason, S.; Garovic, V. Postpartum counseling and interventions to reduce the risk of chronic kidney disease: Back to the future. Kidney Int. 2025, 108, 160–166. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).