1. Introduction
Gene-editing tools such as zinc-finger nucleases, TALENs (transcription activator-like effector nucleases), and the more recent CRISPR-Cas9 system hold significant promise for advancing gene therapy [
1]. The CRISPR-Cas9 technology has enabled precise and easy modifications of the human genome, supporting profound advancements in both research and therapeutic applications. Typically, CRISPR-Cas9 components are delivered into target cells via plasmid DNA-based methods or viral infection systems. The Cas9 endonuclease induces site-specific double-strand breaks (DSBs) guided by single-guide RNAs (sgRNAs), which direct Cas9 to predefined genomic loci [
2]. Mammalian cells respond to DSBs by activating two key repair pathways: non-homologous end joining (NHEJ) and homology-directed repair (HDR) [
3]. NHEJ often introduces random insertions or deletions (indels) at the break site, while HDR leverages homologous DNA sequences, either from homologous chromosomes or donor templates, to precisely introduce or modify sequences at or near the DSB. Such accuracy in homology recombination is critical for precise genome engineering [
4].
However, CRISPR-Cas9-mediated HDR frequently results in monoallelic knock-ins, where only one allele is precisely modified while the other remains unedited or acquires variable indels [
5]. Achieving biallelic knock-in with precise editing is critical when the endogenous expression level of the gene of interest is low from a single allele, or when downstream applications such as functional analyses or screening require a homogeneous population; otherwise, mixed KI/+ and KI/KI cells can confound the interpretation and robustness of the data. Consequently, identifying and selecting cells that have undergone successful biallelic modifications is crucial for various research purposes. Single-cell cloning is a commonly used strategy to identify clones and establish cell lines with monoallelic and/or biallelic gene editing. However, this process is time-consuming and labour-intensive, and is often associated with significant drawbacks, including inherent clonal variability in the parent cells. Additionally, clonally derived reporter cells may not recapitulate the heterogeneity of the parental cells, potentially skewing experimental outcomes. To achieve HDR-directed gene editing, electroporation of plasmid DNA is an efficient transduction approach commonly used for the delivery of sgRNA plasmid and donor DNA into target cells. This delivery method remains challenging to use for many cell types and often requires large amounts of DNA, which can compromise cell viability and lead to substantial random integration [
6]. In addition to this approach, electroporation of pre-assembled CRISPR-Cas9 ribonucleoprotein (RNP) complexes together with donor DNA has emerged as an effective alternative [
7]. Lentiviral vectors present an ideal alternative for delivering gene-editing components due to their high transduction efficiency across diverse cell types. However, conventional lentiviral systems integrate into the host genome, posing risks of insertional mutagenesis and limiting their clinical applicability [
8]. Hence, it is not feasible to deliver a donor DNA template for HDR. Additionally, the continuous expression of gene-editing components in transduced cells increases off-target effects. To address these issues, the integrase-deficient lentiviral vector (IDLV) system has recently emerged as a solution. This system enables transient delivery of sgRNA and donor DNA without genomic integration, minimising off-target and random risks while bypassing the need for electroporation [
9,
10]. Although a few studies support their utilities, further research is needed to fully optimise IDLV-based CRISPR-Cas9 genome editing for the generation of knock-in cells through HDR.
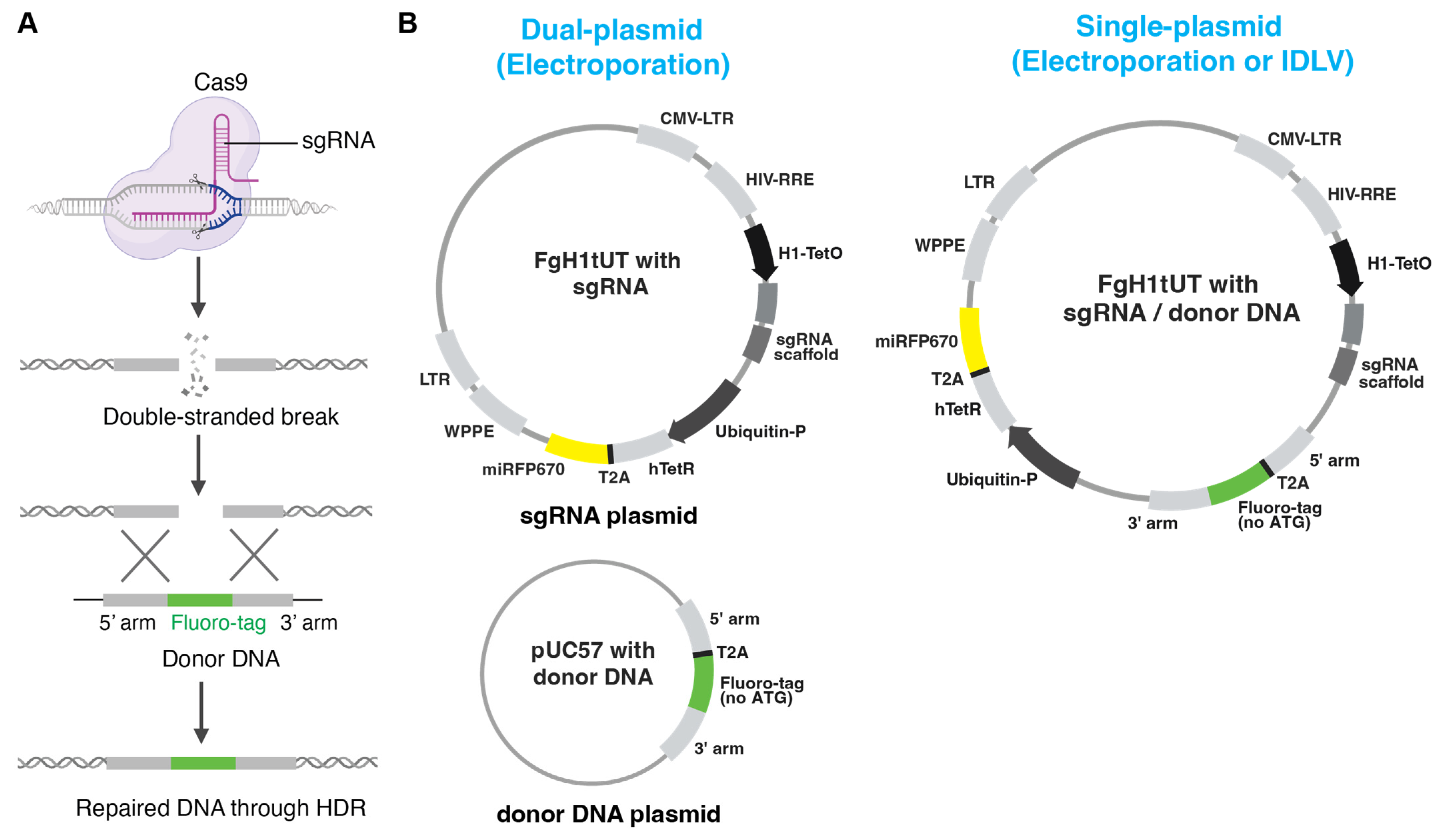
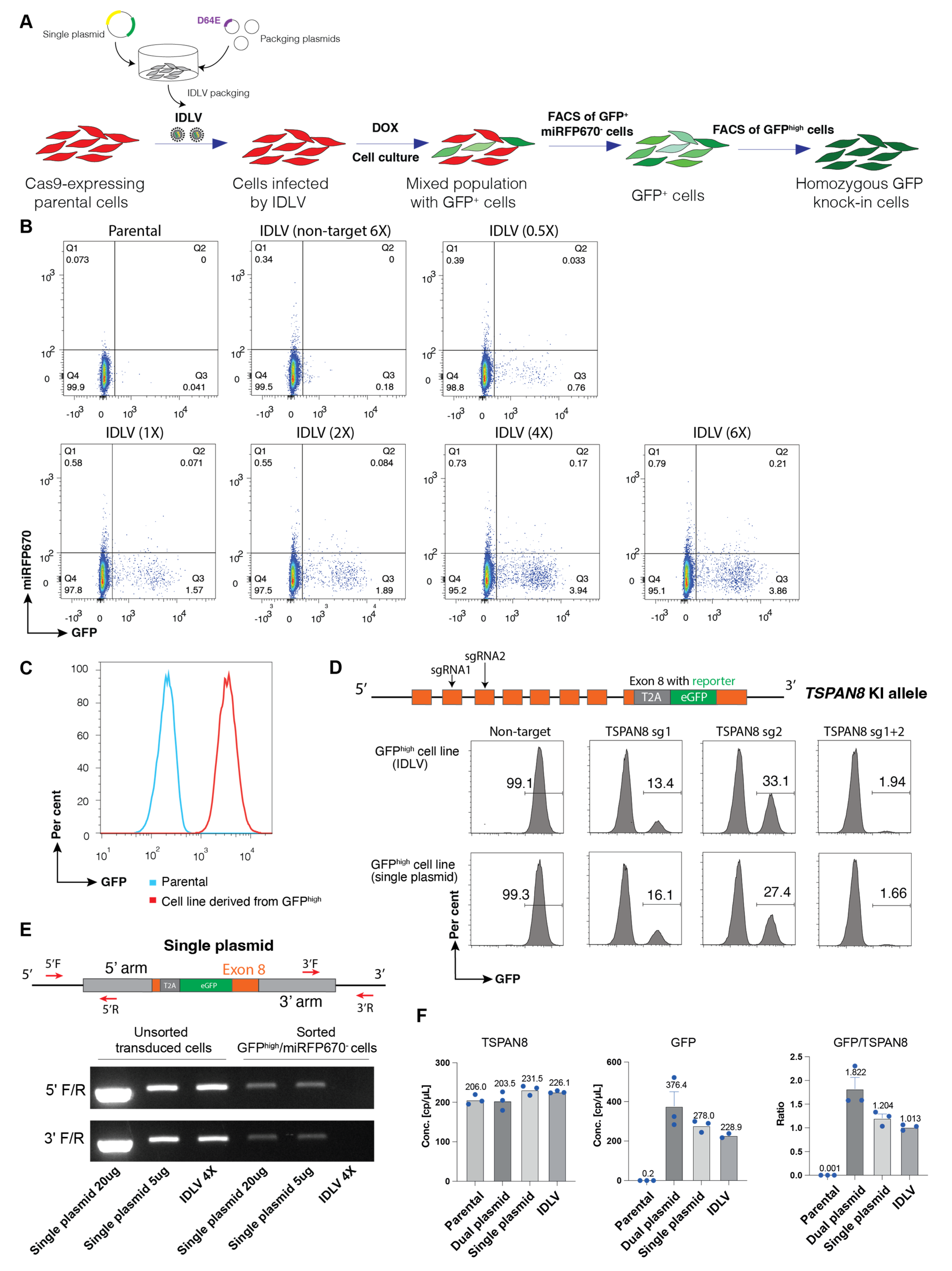
Here, we present and compare three strategies with optimised donor DNA design for generating homozygous fluorescent knock-in cell pools: dual-plasmid and single-plasmid electroporation approaches as well as an IDLV-based delivery system (
Figure 1A,B). The dual-plasmid system employs separate plasmids for donor DNA and doxycycline-inducible sgRNA expression, which are delivered via electroporation. While this approach enables efficient targeted integration of the fluorescent reporter, it is associated with higher rates of random integration. The single-plasmid system integrates both components into one vector, simplifying delivery and incorporating a fluorescent protein marker to eliminate undesired random integration. Especially, the IDLV system showed the lowest random integration while still maintaining high editing efficiency, making it particularly effective for hard-to-transfect cell types. Together, these approaches provide flexible and robust options for rapid generation of homozygous fluorescent reporter knock-in cell pools for a wide range of research purposes.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
Detailed information on all commercially available and validated cell lines utilised in this study can be found in
Table S1. The MEC cell line originated from the pleural effusion of a cholangiocarcinoma patient [
11]. All cell lines were cultured at 37 °C tissue culture incubator with a humidified atmosphere of 5% CO
2. All cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM high glucose, GlutaMAX, ThermoFisher, Waltham, MA, USA, Cat# 10566016) supplemented with 10% (
v/
v) fetal bovine serum (Scientifix, Melbourne, VIC, Australia, Ref: FBSAU-2210C) and 1% (
v/
v) penicillin/streptomycin (ThermoFisher, Waltham, MA, USA, Cat# 15140122), except for JHH5 cells, which were maintained in William’s E media (ThermoFisher, Waltham, MA, USA, Cat# 12551032). Cells were passaged once they reached 80–90% confluency.
2.2. Doxycycline-Inducible sgRNA Lentiviral Vectors
To generate sgRNA plasmids for CRISPR/Cas9 genome editing, the doxycycline-inducible sgRNA vector system Fgh1tUT (Addgene, Watertown, MA, USA, Plasmid #70183) was utilised [
12]. The custom construct Fgh1tUT_miRFP670 was developed in-house by replacing the eGFP gene in Fgh1tUT_GFP with the miRFP670 gene from pmiRFP670-N1 (Addgene, Watertown, MA, USA, Plasmid #79987). The single-plasmid backbone was engineered by introducing multiple cloning sites (MCS, sequence: ACCGGTTGGCGCGCCCCTGCAGGTGCTAGC) into the FgH1tUT_miRFP670 vector to facilitate the efficient subcloning of the donor DNA template into the sgRNA vector. To insert the target sgRNA sequence into a Fgh1tUT vector, two complementary oligonucleotides were synthesised with the following structure: 5′-TCCCNNNNNNNNNNNNNNNNNNNN-3′ and 3′-NNNNNNNNNNNNNNNNNNNNCAAA-5′, then annealed. All specific sgRNA sequences corresponding to the “N” regions are listed in
Table S2. The vector was digested using the BsmBI restriction enzyme. The sgRNAs were either designed using Benchling (
benchling.com/academic) or selected from the Sabatini/Lander CRISPR pooled library (Addgene, Watertown, MA, USA, Cat# 1000000095) as detailed in
Table S2. The
TSPAN8-T2A-GFP donor cassette was synthesised and cloned into a pUC57 vector by a commercial provider. To generate the plasmid for the single-plasmid system, the donor DNA from the pUC57 vector and sgRNA were subcloned into the sgRNA vector with MCS for cloning of donor DNA.
2.3. Transfection of Plasmids into Mammalian Cells by Electroporation
Transient transfections were carried out using the Invitrogen™ Neon™ Electroporation System (Thermo Fisher Scientific, Waltham, MA, USA, Cat# N10025) with the 100 µL Kit, following the manufacturer’s protocol. Briefly, the amount of plasmid DNA was used as indicated in the figures in 100 µL of Resuspension Buffer with 1 million cells. For dual-plasmid transfection, equal donor and sgRNA plasmid DNA were combined to make up the indicated total amount. Electroporation was performed with the following parameters: 870 V pulse voltage, 35 ms pulse width, and two pulses. Immediately after electroporation, cells were transferred to pre-warmed complete culture medium supplemented with 2 µg/mL doxycycline and incubated under standard culture conditions (37 °C, 5% CO2) for 2 days. Following this initial incubation, the medium containing transfection complexes was replaced with fresh complete medium without doxycycline. Cells were then cultured for an additional ~10 days post-transfection to minimise the potential interference of transient donor DNA expression on the analysis.
2.4. Production of Integrase-Competent Lentivirus (ICLV) and Infection
Lentiviral particles were generated via transient transfection of HEK 293 cells cultured in 10 cm dishes at 70% confluence using 3 μg of target plasmid DNA, 3 μg of pMDLg/pRRE, 2 μg of pMD2.G, and 2 μg of pRSV-Rev. Virus-containing supernatants were harvested 48 h post-transfection and filtered through a 0.45 μm membrane. The filtered supernatants supplemented with 2 μg/mL polybrene were added to target cells and centrifuged at 500× g for 30 min. Following 24 h incubation under standard culture conditions (37 °C, 5% CO2), the infected cells were cultured in fresh complete medium.
2.5. IDLV-Based Infection for Inducing Reporter Knock-In
Lentiviral particles were produced by transiently transfecting HEK 293 cells cultured in 10 cm dishes at 70% confluence with 3 μg of lentivirus plasmid DNA, 3 μg of pBK43 (psPAX2-D64E), 2 μg of pMD2.G, and 2 μg of pRSV-Rev. Virus-containing supernatants were collected 48 h post-transfection and filtered through a 0.45 μm membrane. The viral supernatants, supplemented with 2 μg/mL polybrene and 2 μg/mL doxycycline, were added to cultured cells, followed by centrifugation at 500× g for 30 min. Notably, doxycycline was added simultaneously with the viral supernatant to promptly induce sgRNA expression. After 48 h of incubation under standard culture conditions (37 °C, 5% CO2), the infected cells were re-cultured in fresh medium. Cells were then cultured for an additional ~10 days post-transfection to allow for knock-in stabilisation. To determine the multiplicity of infection (MOI), target cells were plated and infected with serial dilutions of IDLV virus expressing the miRFP670 reporter. After 48 h, cells were harvested and analysed by flow cytometry to assess miRFP670 expression. MOI = 0.5 was defined as the condition where around 50% of cells were miRFP670-positive.
2.6. Flow Cytometry Analysis and FACS Sorting
For FACS analysis of cell-surface TSPAN8 expression, an APC-conjugated anti-human TSPAN8 antibody was used (REAfinity™, Miltenyi Biotec, Bergisch Gladbach, Germany, Clone REA443, Cat# 130-106-811). Prior to analysis, cells were stained with 0.2 μg/mL 7-AAD (Caymanchem, Ann Arbor, MI, USA) to exclude dead cells. Flow cytometry analysis was conducted using the Fortessa cell analyser (Becton Dickinson, Franklin Lakes, NJ, USA), and cell sorting was carried out on the FACS Aria (Becton Dickinson, Franklin Lakes, NJ, USA). Data were analysed using FlowJo software (v10, Tree Star, Ashland, OR, USA).
2.7. Western Blot Analysis
The primary antibodies used for Western blotting are listed in
Table S1. Cells were lysed in ice-cold cell lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.3, 0.25 mM EDTA, pH 8.0, 1% sodium deoxycholate, 1% Triton X-100, 0.2% sodium fluoride and 0.1% sodium orthovanadate), supplemented with a protease and phosphatase inhibitor cocktail (Roche, Mannheim, Germany). The lysates were separated on 10% SDS-PAGE and transferred to PVDF membranes. After blocking with 5% non-fat milk in wash buffer (TBS, 0.01% Triton X-100, Sigma-Aldrich, Merck Pty Ltd, Castle Hill, NSW, Australia) at 37 °C for 1 h, immunoblotting was performed by incubating the membrane with primary antibodies overnight at 4 °C. The membrane was then washed three times with wash buffer and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualised using the Odyssey CLx (Image Studio 3.1 software; LI-COR Biosciences, Lincoln, NE, USA), as per the manufacturer’s instructions.
2.8. Polymerase Chain Reaction (PCR) Genotyping
Confirmation of the correct integration in the reporter cell lines was performed by PCR genotyping. The genomic DNA were extracted as per the manufacturer’s instructions (One-4-All Genomic DNA Miniprep Kit, Cat# BS88504, Bio Basic, Markham, ON, Canada). The PCRs were carried out on a T100 thermocycler (BioRad, Hercules, CA, USA) using the following settings: initial denaturation at 95 °C for 3 min; 28 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 45 s, and extension at 72 °C for 1 min. A final extension step was performed at 72 °C for 5 min, followed by a hold at 4 °C until further use. The specific PCR primers used are listed in
Table S3.
2.9. Genome-Wide CRISPR/Cas9 Knockout Screening
The Human Two Plasmid Activity-Optimized CRISPR Knockout sgRNA Library, developed by David Sabatini, Eric Lander and colleagues, was obtained from Addgene (Cat# 1000000095, Watertown, MA, USA) [
13]. Lentiviral particles were produced by packaging the library into HEK 293 cells. Pilot experiments were performed to determine the virus input required to achieve MOI = 0.3–0.5, corresponding to around 70% and 50% cell death upon puromycin selection, respectively, as previously described [
14]. For screening, cells were seeded in T150 flasks and cultured prior to infection. Approximately 300 million cells were exposed to lentivirus in a medium supplemented with 2 μg/mL polybrene at an MOI of 0.3–0.5 for two days. Infected cells were then selected with 10 µg/mL puromycin in fresh media for two days and subsequently cultured in puromycin-free fresh media for several days before FACS sorting. A total of 10–20% of the total infected cells were reserved as the pool control sample, and the remaining 80–90% of cells were used for FACS sorting based on GFP expression. The genomic DNA was extracted from both the pooled control and sorted cells according to the manufacturer’s instructions (Bio Basic, One-4-All Genomic DNA Miniprep Kit, Markham, ON, Canada). NGS libraries were generated via PCR using specific barcode primers. The sgRNA barcode PCR primers we as follows: Forward: CAAGCAGAAGACGGCATACGAGATCnnnnnnTTTCTTGGGTAGTTTGCAGTTTT; Reverse: AATGATACGGCGACCACCGAGATCTACACnnnnnnnnCACCGACTCGGTGCCACTTTT. “n” represents sample-specific barcode sequences. Multiplexed NGS libraries were sequenced on the HiSeq4000 platform (Illumina, San Diego, CA, USA) to identify the sgRNAs in each sample. Data normalisation and sgRNA modelling and ranking were performed by using the MAGeCK algorithm as previously described [
15].
2.10. Generation of CRISPR-Cas9 Knockout Cells
To generate sgRNA-induced gene knockout cells, a two-plasmid lentiviral sgRNA inducible expression system was employed, which consists of (1) FuCas9Cherry vector (Addgene#70182, Watertown, MA, USA), which constitutively expresses Cas9 and the mCherry fluorescence marker, and (2) FgH1tUT vector, which drives doxycycline-inducible sgRNA expression along with constitutive expression of either eGFP or TagBFP, as previously described. Cells were first transduced with the pFU_Cas9_mCherry plasmid and sorted using the FACS Aria (Becton Dickinson, Franklin Lakes, NJ, USA) to establish the stable Cas9_mCherry expressing populations. The Cas9 stable cells were then infected with lentivirus carrying doxycycline-inducible sgRNAs (Fgh1tUT vectors) targeting coding exons of the gene of interest. To activate the sgRNA expression via the Tet-O promoter, cells were treated with 1ug/mL doxycycline at 37 °C in a humidified incubator with 5% CO2 for two days. Following that, cells were washed with PBS and cultured in a fresh and doxycycline-free complete medium for a minimum of five days. KO populations were enriched by FACS, selecting the cells double positive for GFP and BFP.
2.11. Reverse Transcription and qPCR Analysis
Total RNA was extracted following the manufacturer’s protocol (Qiagen, RNeasy Micro Kit, Cat# 74004, Hilden, Germany). mRNA was reverse-transcribed into cDNA with GoScriptTM Reverse Transcription Kit (Promega, Cat# A5000, Madison, WI, USA). The resulting cDNA was used for either gene cloning via PCR amplification or for qPCR. qPCR was performed using SYBR Green qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) with specific forward and reverse primers. Reactions were run on a Bio-Rad System (Bio-Rad, Hercules, CA, USA). Relative mRNA expression levels were calculated by the standard delta-delta Ct (2−∆∆Ct) method, with β-actin serving as the housekeeping control.
The TSPAN8 primers used for qPCR were as follows: Forward: GCAGAGACCATGCCAAAGCTATAATG and Reverse: CGATCTGGCAATACAGGACCATAG. β-actin primers used for qPCR; Forward: TCCCTGGAGAAGAGCTACG and Reverse: GTAGTTTCGTGGATGCCACA.
2.12. Long-Range PCR for TSPAN8
Genomic DNA was extracted using the Qiagen DNeasy Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Long-range PCR amplification of the TSPAN8 locus was performed using Takara PrimeSTAR GXL DNA Polymerase (Cat# R051A, Takara Bio, Kusatsu, Shiga, Japan). PCR cycling conditions were as follows: 95 °C for 2 min, followed by 30 cycles of 95 °C for 30 s and 68 °C for 8 min, with a final extension at 68 °C for 5 min. Two primer sets for the TSPAN8 genomic region spanning the sgRNA targeting site were used: Set 1—forward primer F1: 5′-GTCTATAACCTGCCCTCCCTCTTTTTAAGG-3′ and reverse primer R1: 5′-GCAAAAGAAACTACCATCAGAGTGAACAGG-3′; Set 2—forward primer F5: 5′-TGAATGACCTCTCTACCGGGAAAAAGATAG-3′ and reverse primer R5: 5′-CATAACGAAATGAAGGCAGAAATGAAGATG-3′. PCR products were analysed by agarose gel electrophoresis.
2.13. Droplet Digital PCR (ddPCR)
Digital PCR was performed using the Qiagen QIAcuity Four system with genomic DNA extracted using the Qiagen DNA extraction kit (Qiagen, Hilden, Germany) and quantified by the high-sensitivity dsDNA quantification kit for Qubit (Invitrogen, Waltham, MA, USA). The same amount of genomic DNA for each sample was digested with the 4-cutter restriction enzyme AluI and carried forward into the ddPCR reaction. For detection of eGFP integration, the following primers and probe were used: forward primer GACAACCACTACCTGAGCAC, reverse primer CAGGACCATGTGATCGCG, and probe CCTGAGCAAAGACCCCAACGAGAA labelled with 6-FAM (5′), ZEN (internal quencher), and Iowa Black FQ (3′). For quantification of the upstream 5′ homology arm of the human TSPAN8 locus, the assay included forward primer GATGATCAGCACTTTCCTTGC, reverse primer ACTGCTTTATTTCTAGGACCTCC, and probe TGATGGCTCTCAGTGTGTAGCACTTTT labelled with Texas Red-X (5′) and Iowa Black RQ-Sp (3′). Thermal cycling was conducted using the standard probe-based duplex protocol provided by Qiagen (Hilden, Germany).
2.14. Quantification and Statistical Analysis
Unless otherwise specified, data are presented as mean ± standard error of the mean (SEM), and the significance was assessed by unpaired t-test using GraphPad Prism (v10, GraphPad Software, San Diego, CA, USA), with p < 0.05 considered significant.
4. Discussion
CRISPR-Cas9 genome editing has revolutionised the ability to engineer specific loci in cultured cells. However, the laborious process of isolating clonal populations with desired modifications remains a major bottleneck. This problem becomes even more challenging when attempting to generate homozygous knock-in reporter cell lines via HDR, where low biallelic editing efficiencies typically yield predominantly heterozygous clones. In some cases, two rounds of iterative CRISPR tagging may be necessary to achieve homozygosity. In this study, we present optimised strategies for rapid and robust generation of homozygous fluorescent knock-in reporter cell pools using CRISPR-Cas9, which are particularly valuable for studying gene regulation and function, especially in cancer cell models where inherited heterogeneity poses a key challenge.
Our approach is based on the principle that cells with biallelic knock-ins display roughly twice the fluorescence intensity of those with monoallelic knock-ins, enabling reliable discrimination between the two populations. the fluorescent reporter intensity in cells with biallelic knock-in is approximately twice that of cells with monoallelic knock-in. Hence, a pool of the rare homozygous fluorescent knock-in clones can be rapidly isolated by FACS from a large population of the target cells to establish a homozygous knock-in reporter cell line. Eliminating the background expression of the fluorescent reporter from random integration is crucial for the success of the protocol. To ensure the expression of GFP is only from the locus with correct integration of the donor DNA through HDR, we used the T2A system and removed the start codon of the fluorescent reporter in the donor DNA design. Moreover, we utilised a doxycycline-inducible system to transiently switch on the expression of sgRNA. The data clearly demonstrate tight doxycycline control for both plasmid electroporation and IDLV-mediated delivery: successful gene editing, indicated by the expression of reporter, occurred only in the presence of doxycycline (
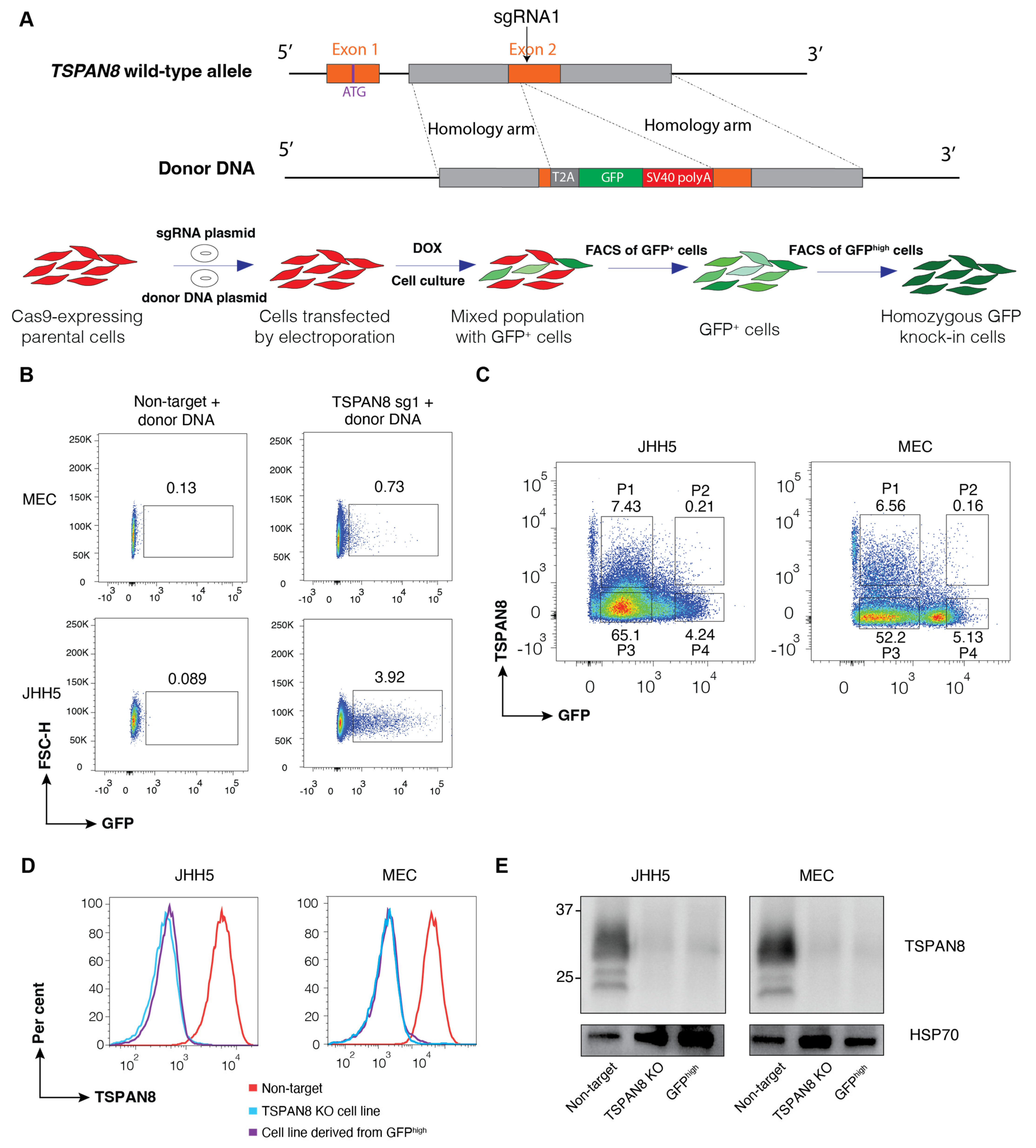
Figure S2A). We validated the feasibility and efficiency of our strategy by generating TSPAN8 knockout cells, using knock-in GFP reporter expression as a surrogate, through co-transfection of two separate plasmids encoding the sgRNA and donor DNA via electroporation. Our results clearly show that cell clones with precise biallelic integration of GFP can be identified and isolated by FACS based on higher expression of the knock-in reporter. It is important to note that the large proportion of GFP
low cells also exhibited a complete absence of the TSPAN8 protein. However, unlike the GFP
high knockout cells, which underwent precise genomic editing, these GFP
low cells likely harbour a large variety of out-of-frame indels in the second allele, which is not ideal for certain downstream utilities of knock-out cells.
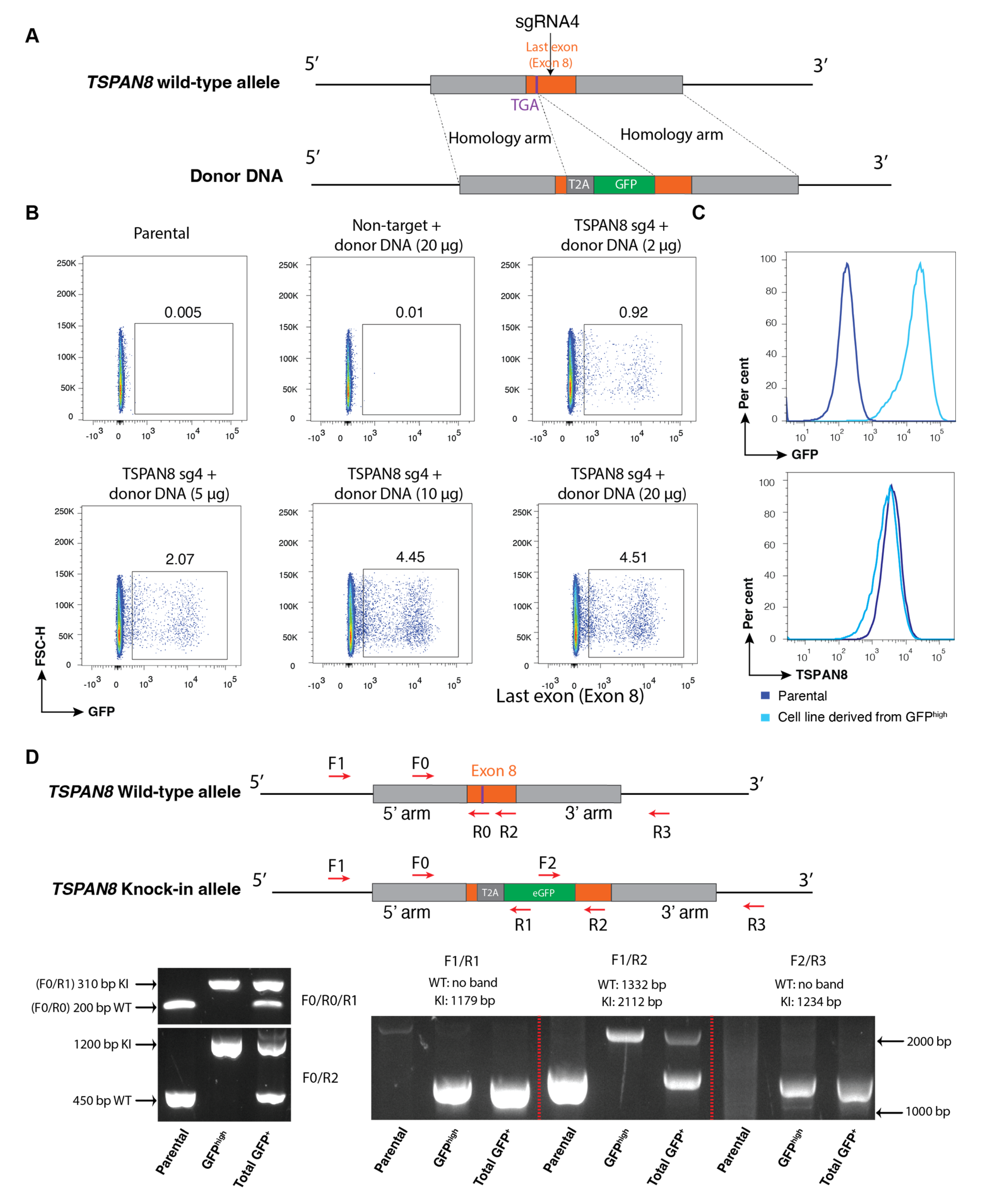
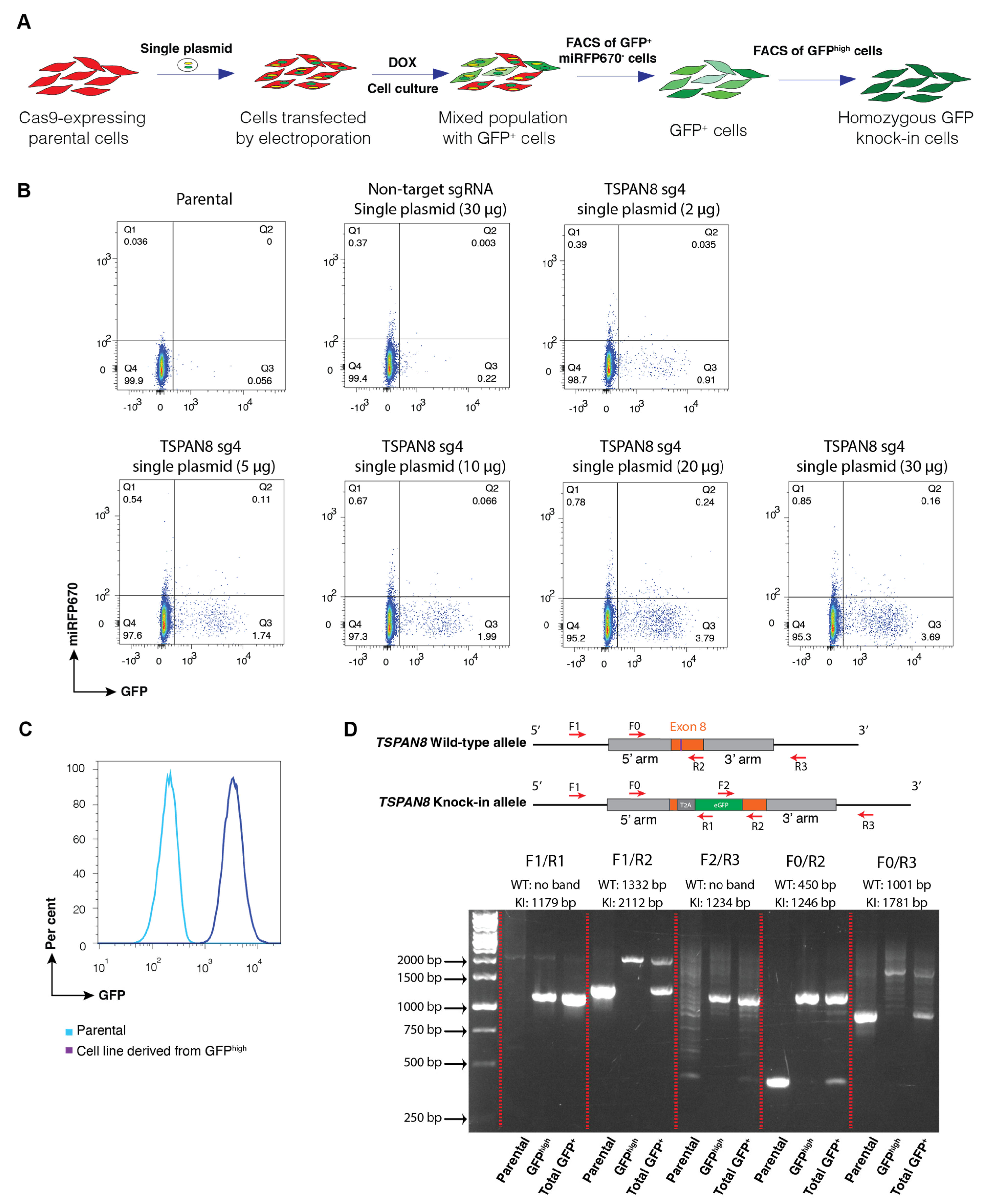
We next applied a similar approach to generate knock-in reporter cell pools by replacing the stop codon of a target gene with the T2A-GFP cassette, thereby preserving the endogenous transcriptional regulation of the target gene as much as possible. For these cell lines, we confirmed successful biallelic knock-in using PCR genotyping with multiple specific primer sets. To further streamline our method, we developed a single-plasmid system in which both the sgRNA expression cassette and donor DNA template for HDR are incorporated into a single vector. Our data demonstrated that sgRNA was successfully induced to mediate the cleavage of the desired gene locus, while the plasmid served as the donor DNA for the efficient integration of the reporter through HDR. The single-plasmid electroporation system lowers the risk of random integration by incorporating the miRFP670 reporter, allowing exclusion of undesired cells; however, it exhibits slightly lower HDR efficiency compared to the dual-plasmid system.
It is well known that certain cell types are resistant to transfection or electroporation, and often exhibit high levels of cell death following these procedures. In contrast, the lentivirus infection as a tool for the delivery of genetic materials shows high efficiency and low cytotoxicity across a wide range of cancer and primary cells. However, lentivirus generated using the conventional packaging system is not feasible for transient expression of sgRNA and delivery of donor DNA for HDR due to the frequent integration of virus-derived DNA into the host genome following infection. To overcome this challenge, we leveraged the non-integrating nature of IDLV, due to a lack of a functional integrase enzyme, and combined it with our single-plasmid system to develop a novel approach for generating reporter knock-in cells by CRISPR-Cas9 genome editing. Remarkably, we found that the IDLV-based approach enables the generation of homozygous knock-in reporter cells with a similar efficiency to the single-plasmid electroporation. Despite optimised plasmid design and the incorporation of a constitutively expressed fluorescent reporter to minimise cells with random integration, PCR genotyping still revealed instances of unintended donor plasmid integration into the genome. While this may not significantly impact the utility of the established knock-in reporter cells for many applications, the cell lines generated using the IDLV approach exhibited minimal or undetectable levels of random integration. In line with this, ddPCR analysis confirms no alternation in the copy number at the target locus across all the approaches, and demonstrates a consistent 1:1 copy number ratio between the reporter and the TSPAN8 gene, strongly suggesting that random integration via the IDLV method is negligible. We speculate that this is likely due to the substantially lower amount of donor DNA delivered into cells when using the IDLV system, as compared to the high DNA load introduced by electroporation.
Our approaches entirely rely on FACS sorting according to the expression levels of the correctly integrated knock-in fluorescent reporter driven by the endogenous promoter of the target gene. Hence, they are not suitable for generating knock-in cells expressing non-fluorescent protein reporters, such as LacZ, luciferases, or other enzymes. Compared to clonal fluorescent knock-in reporter lines established by previous strategies, homozygous knock-in cell pool lines engineered by our methods significantly eliminate the issue associated with inherited clonal variations within the parental cells. However, our approaches require the target gene’s endogenous expression to be detectable and sufficiently uniform. Accordingly, one key limitation is that the methods developed here are not feasible for generating reporter knock-in cells for the genes that are silent in the parent cells. While the design of the donor DNA and use of the IDLV-based delivery system profoundly reduce random integration and background expression of the knock-in reporter, this study does not address issues related to the off-target effect of CRISPR-Cas9 genome editing. Alternative CRISPR-Cas9 genome editing approaches are steadily advancing. One effective method involves electroporating Cas9 ribonucleoprotein (RNP) complexes along with donor plasmids, which has demonstrated high editing efficiency [
30]. Another strategy employs adeno-associated virus (AAV) vectors to deliver single-stranded DNA templates [
31]. The approaches developed in this study offer an additional, valuable and distinctive option. Finally, it is important to note that our study is dependent on HDR for knock-in efficiency, which can vary across different cell types and phases of the cell cycle, and some cells may preferentially utilise NHEJ, limiting the generalizability of our method in settings where HDR is less active, such as cells with a defect in BRCA1 function.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}