1. Introduction
Parkinson’s disease (PD), the second most common neurodegenerative disorder, is classically defined by the progressive degeneration of nigrostriatal dopaminergic neurons and subsequent motor impairments [
1,
2]. While neuronal pathology has dominated PD research, emerging evidence implicates oligodendrocyte dysfunction as a critical contributor to disease progression [
3,
4]. These glial cells not only maintain axonal integrity via myelination but also support neuronal survival through metabolic coupling. Clinical observations reveal that PD patients often exhibit white matter lesions (WMLs) in cortical regions, which correlate with the severity of motor symptoms [
5]. Furthermore, recent whole-brain connectivity omics analyses have demonstrated reduced myelin content in the brains of PD patients, particularly in circuits associated with dopaminergic neurons [
6], indicating that myelin disruption may impair dopaminergic signaling [
7,
8]. However, the molecular mechanisms driving oligodendrocyte impairment in PD remain elusive.
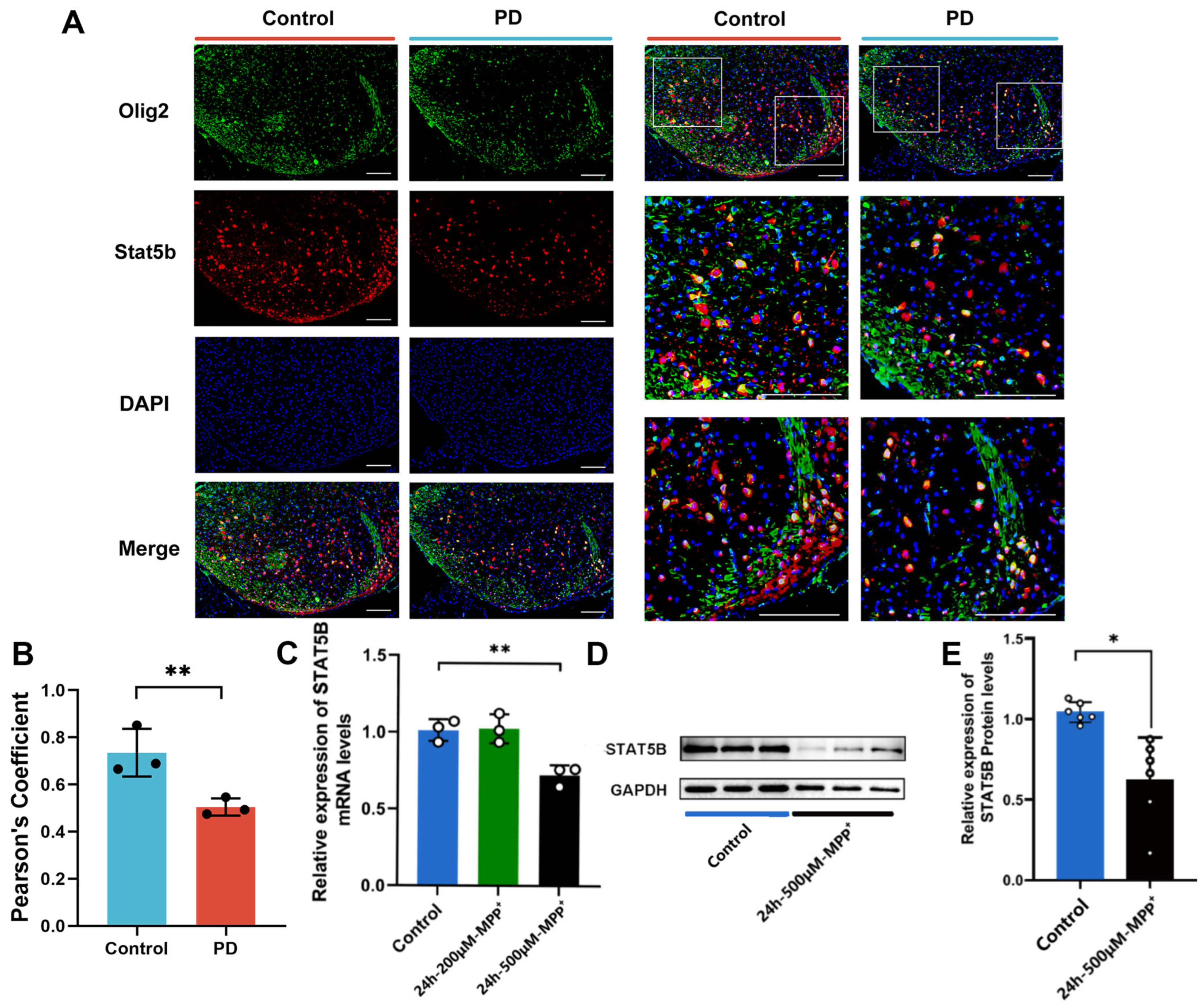
Signal transducer and activator of transcription 5B (STAT5B), a pleiotropic transcription factor regulating cell survival and differentiation [
9,
10], has demonstrated neuroprotective effects in neurodegeneration [
11,
12,
13]. In spinal muscular atrophy, STAT5 activation preserves motor neuron function [
14]. STAT5B deficiency reduces striatal dopamine levels and exacerbates dopaminergic neuron loss [
15]. Critically, our reanalysis of single-nucleus RNA sequencing (snRNA-seq) data from an MPTP-induced mouse model revealed the selective downregulation of
STAT5B transcriptional activity in substantia nigra (SN) oligodendrocytes, with STAT5B expression positively correlated with myelin integrity. However, the mechanism by which oligodendrocytic STAT5B regulates myelin maintenance and dopaminergic neuron survival in PD remains unclear.
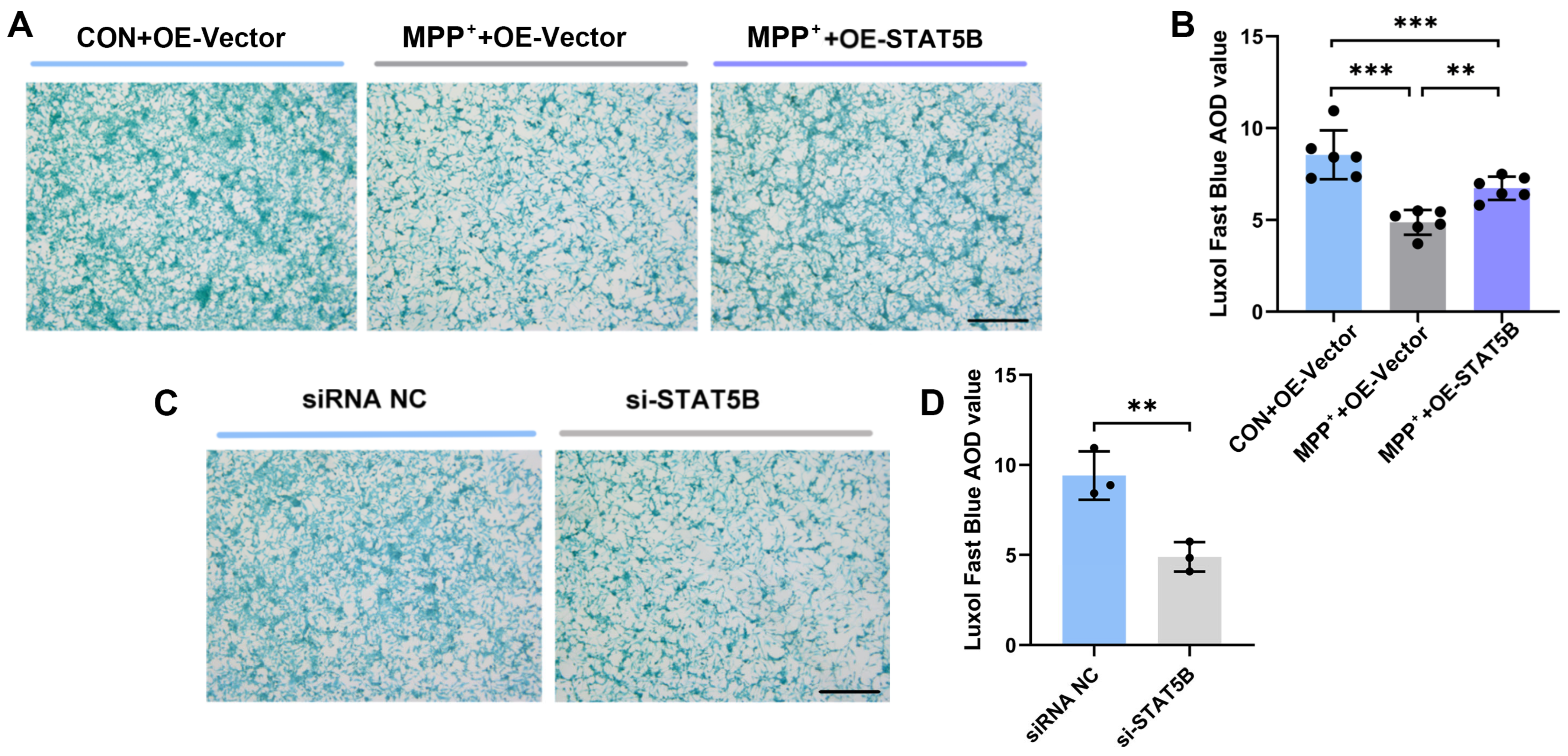
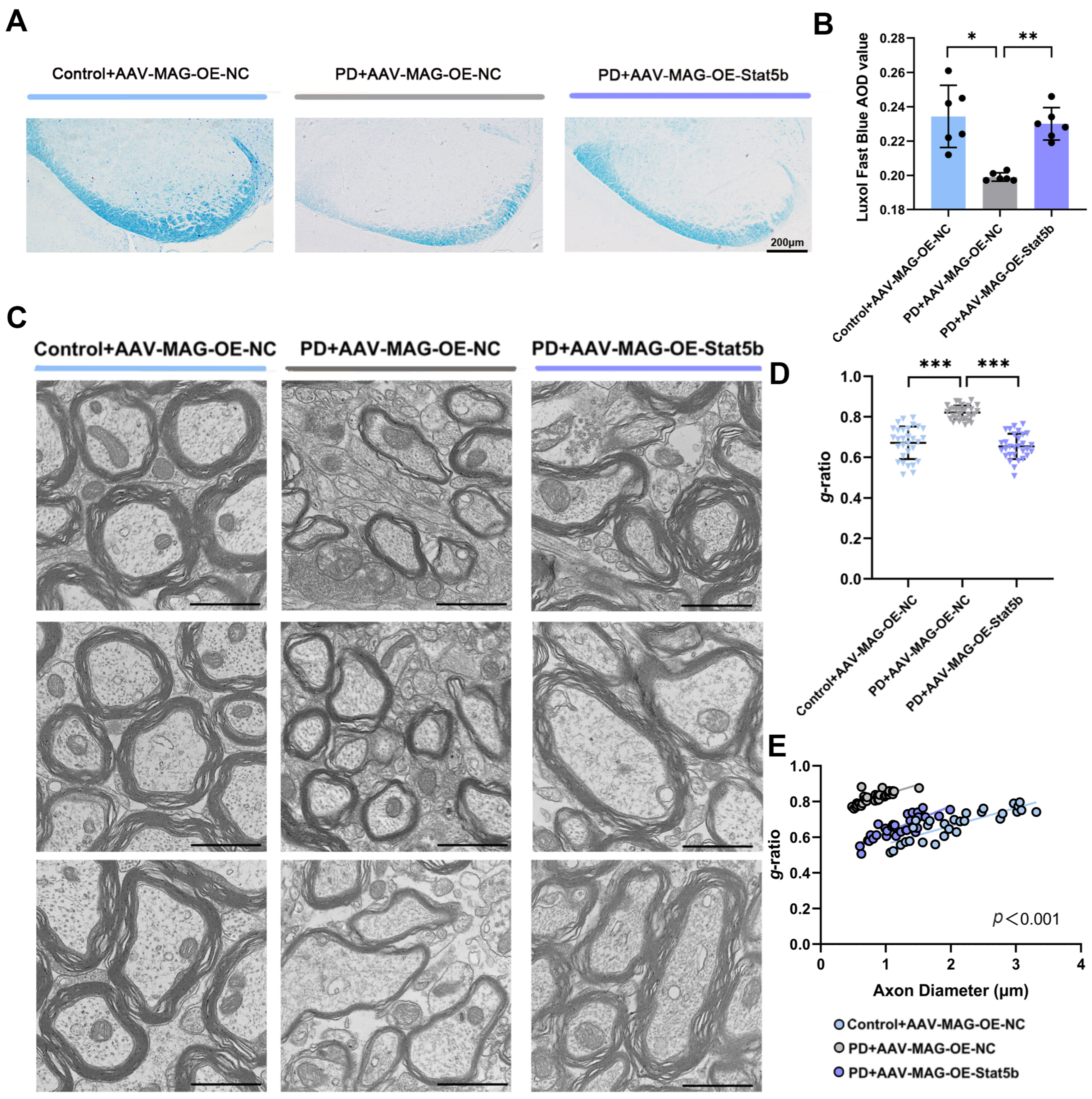
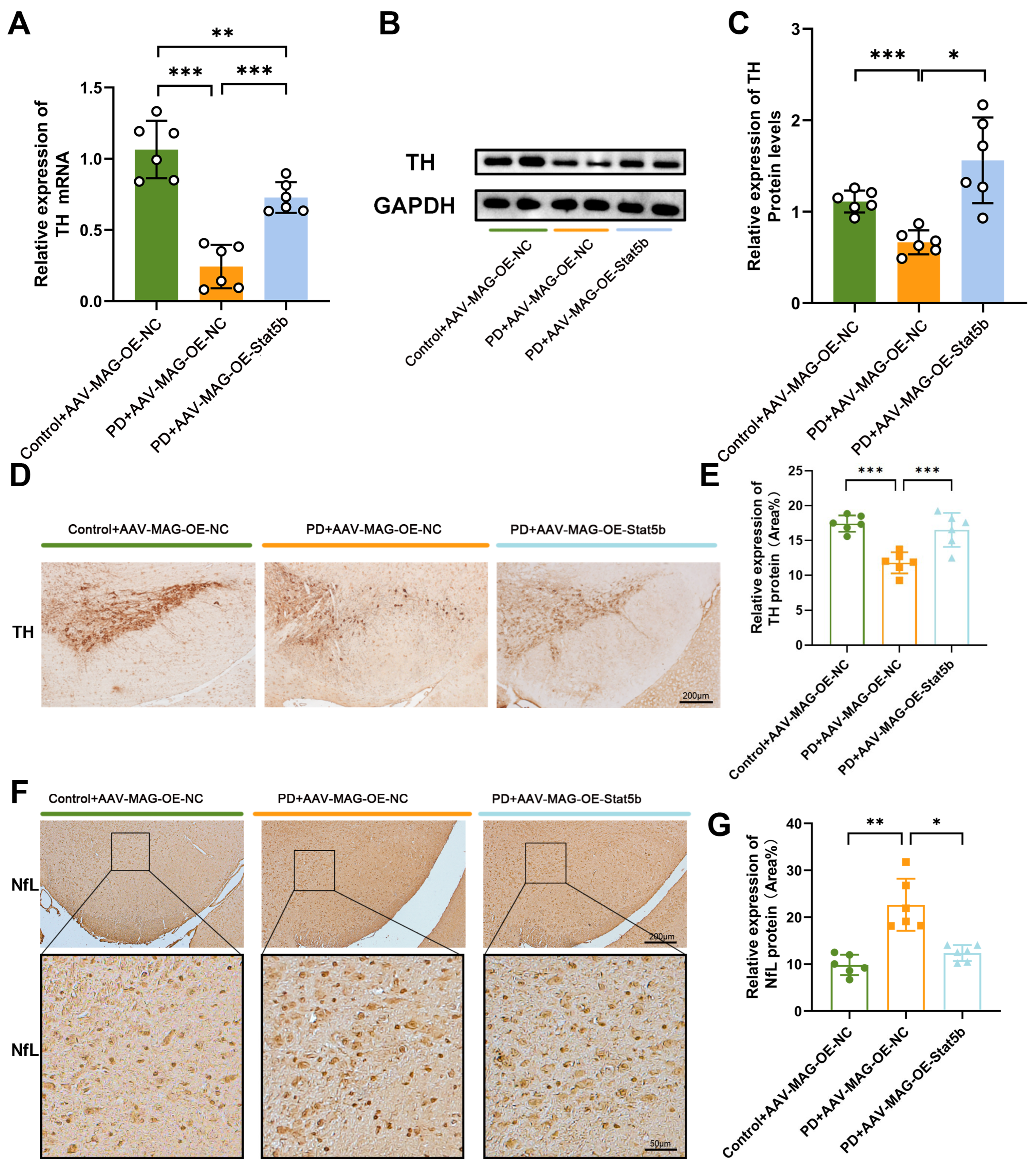
In this study, we demonstrate that restoring STAT5B expression in oligodendrocytes rescues myelination defects and motor dysfunction in PD models. Mechanistically, STAT5B contributes to myelin damage by transcriptionally inhibiting myelin basic protein (MBP) expression. Furthermore, we elucidate DNA methyltransferase 3A (DNMT3A)-mediated hypermethylation at the STAT5B promoter (position 2202) as the key epigenetic mechanism underlying decreased STAT5B transcription in PD.
By uncovering the DNMT3A-STAT5B-MBP axis in PD pathogenesis, we reveal how oligodendrocyte epigenetic dysregulation drives dopaminergic circuit failure, propose STAT5B reactivation as a dual-target therapy for myelin repair and neuroprotection, and nominate STAT5B promoter methylation as a translatable progression biomarker.
2. Materials and Methods
2.1. Establishment of MPTP-Induced Mouse Model
To establish an animal model of PD, 8-week-old male C57BL/6 mice were intraperitoneally (i.p.) administered 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Macklin, catalog number: M832929, Shanghai, China) at a daily dose of 30 mg/kg body weight for five consecutive days (PD group). The control group received an equivalent volume of physiological saline. Behavioral assessments were performed to evaluate and compare the physical activity levels of both groups. All animal procedures were conducted in accordance with the Animal Welfare Act, as well as the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996), and received approval from the ethics committee at Hebei Medical University (IACUC-Hebmu-P-2023157). Animals with pre-existing health conditions were excluded from the study. All researchers involved in the study were blinded to group assignments.
2.2. Mouse snRNA-Seq Data Processing
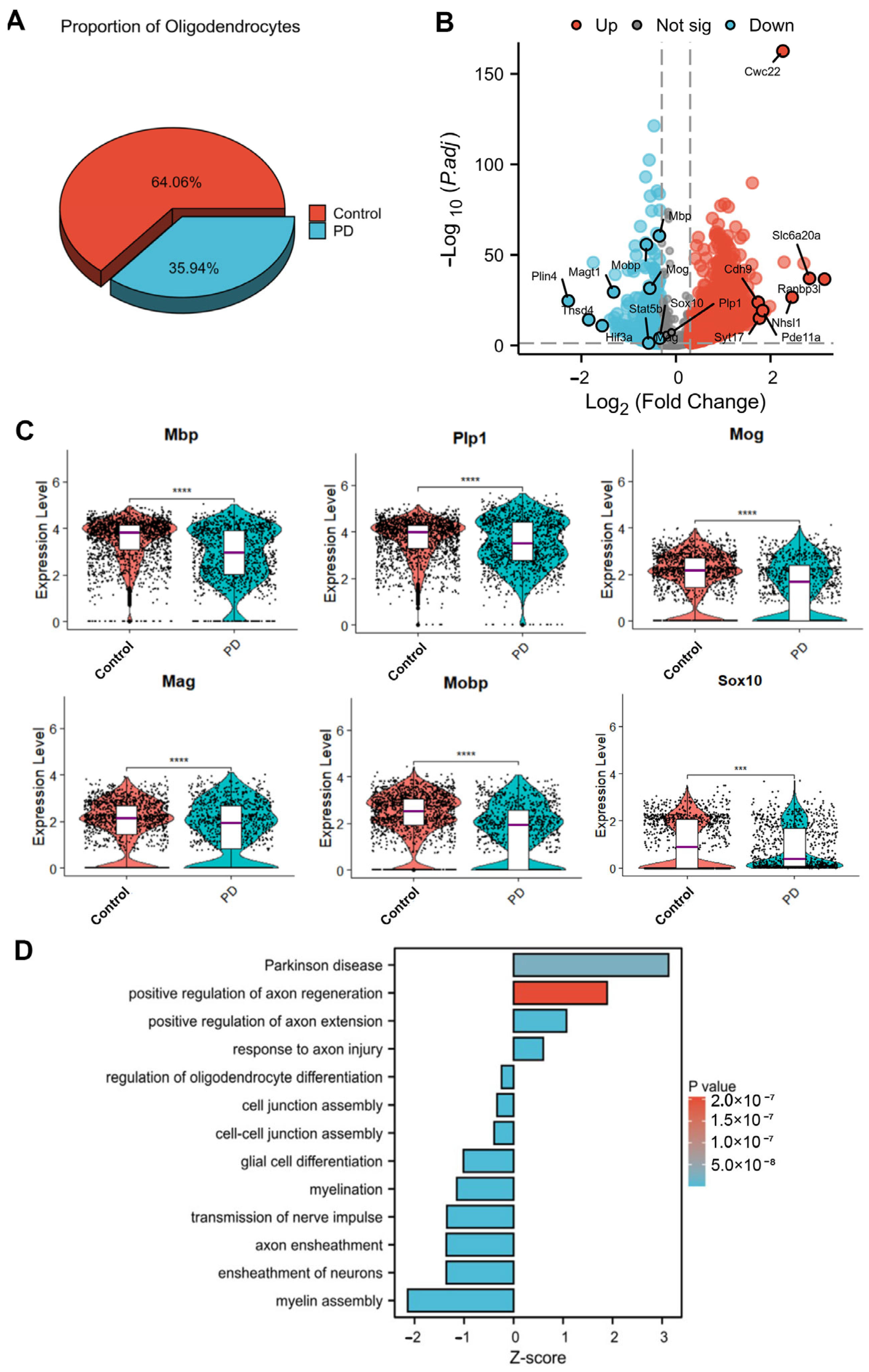
The data were processed using Seurat (v4.2.2) following standard pipelines. After quality control (excluding cells with >20% mitochondrial genes), we performed uniform manifold approximation and projection (UMAP)-based clustering (resolution = 0.5) to identify 25 initial clusters. Cell types were annotated using marker genes (identified by FindAllMarkers function) and cross-referenced with the CellMarker and PanglaoDB databases. Oligodendrocytes were subsequently subclustered for detailed analysis. For differentially expressed gene (DEG) analysis, we compared the MPTP-induced PD and control groups using FindMarkers (thresholds: p < 0.05, |log2FC| > 0.5). Significant DEGs in oligodendrocytes underwent functional enrichment analysis via clusterProfiler (v3.18.4), with false discovery rate (FDR) correction for multiple testing.
2.3. Analysis of the Whole Mouse SN Dataset
2.3.1. Inference of Pseudotime Ordering Using Monocle2
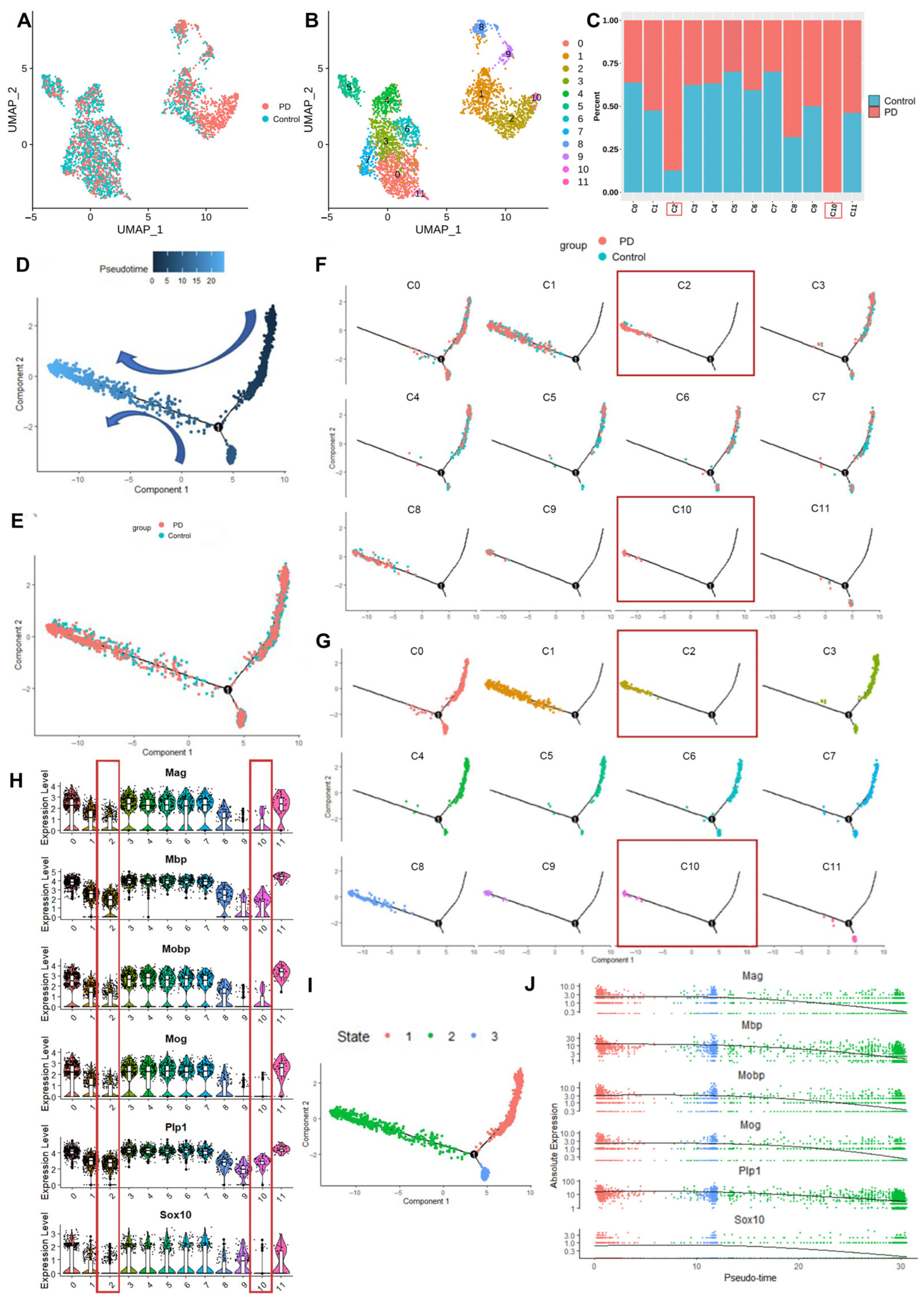
To elucidate alterations in oligodendrocyte cellular states between the PD and control groups, we employed Monocle2, a robust algorithm for the reconstruction of cellular state trajectories after subclustering oligodendrocytes. This analysis critically involved identifying genes that potentially influenced the observed cellular state changes between the PD and control groups and their associations with the cellular state trajectory.
2.3.2. DoRothEA-Predicted Regulon Activity
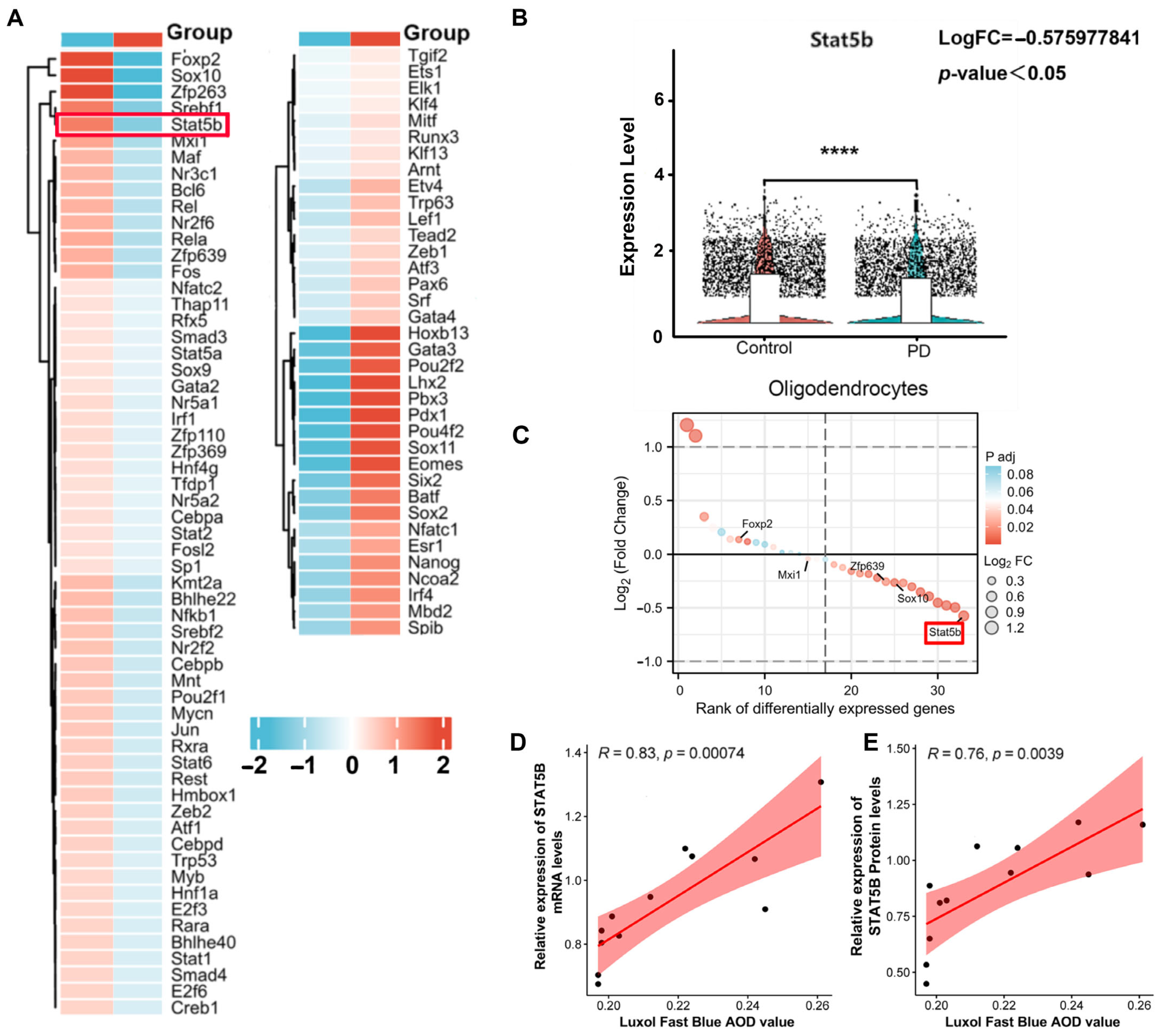
To explore changes in transcription factor (TF) activity in oligodendrocytes between the PD and control groups, we utilized the DoRothEA (v3.21) R package [
16]. DoRothEA is a gene regulatory network (GRN) resource that deduces TF activity from gene expression data by analyzing TF–target gene interactions. We applied DoRothEA with confidence levels A, B, and C to compute viper scores, which quantified differential TF activity between the PD and control groups.
2.4. Mouse Brain Stereotactic Injection of the SN
Mice were anesthetized with isoflurane and immobilized in a stereotactic frame. After scalp disinfection and incision, we targeted the SN using coordinates relative to bregma and lambda (AP: −3.11 mm, ML: ±1.4 mm, DV: −4.2 mm). Adeno-associated virus (AAV) was delivered through a drilled craniotomy, with the needle maintained for 5 min post-injection before gradual withdrawal. The scalp was sutured and mice received standard postoperative care. Viral expression and behavioral outcomes were assessed 21 days post-injection.
2.5. Immunofluorescence Staining
Immunofluorescence was performed on cultured cells and 10 μm frozen substantia nigra (SN) sections. After PBS washes, sections were blocked with 5% donkey serum in PBS-T (0.5% Triton X-100) for 60 min at room temperature. Primary antibody incubations (4 °C overnight) included the following: Olig2 mouse monoclonal (1:300, sc-515947, Santa Cruz Biotechnology, Shanghai, China), STAT5B rabbit (1:200, CSB-PA022815LA01HU, CUSABIO, Wuhan, China) and mouse (1:200, 66427-1-Ig, Proteintech, Wuhan, China) monoclonals, and MBP rabbit monoclonal (1:200, 10458-1-AP, Proteintech, Wuhan, China). Sections were then incubated with Alexa Fluor-conjugated secondary antibodies (1:500, Thermo Scientific™, Shanghai, China) for 2 h: donkey anti-rabbit IgG-Alexa Fluor 594 (A-21207), donkey anti-mouse IgG-Alexa Fluor 488 (A-21202). After DAPI mounting, images were acquired using an Olympus FV1200 microscope (Olympusm, Tokyo, Japan) and analyzed with ImageJ (v1.51).
2.6. Immunohistochemical Staining
Mouse SN paraffin sections (5 μm thick) underwent dehydration through an ethanol series, followed by antigen retrieval using citrate buffer to facilitate immunohistochemical staining. Section processing strictly followed the manufacturer’s guidelines, as outlined in the UltraSensitiveSP Kit (catalog number: KIT-9720, MXB Biotechnologies, Fuzhou, China). To inhibit endogenous peroxidase activity, sections were treated with 0.3% hydrogen peroxide and subsequently incubated with fetal bovine serum (FBS) at 37 °C for 30 min. Following this, sections were incubated overnight at 4 °C with an anti-TH rabbit monoclonal antibody (1:200, catalog number: PB9449, Boster, Wuhan, China) and an anti-NfL mouse monoclonal antibody (1:300, catalog number: sc-71678, Santa Cruz Biotechnology, Shanghai, China). Image analysis was performed using the Image-Pro Plus 6.0 software.
2.7. Luxol Fast Blue Staining
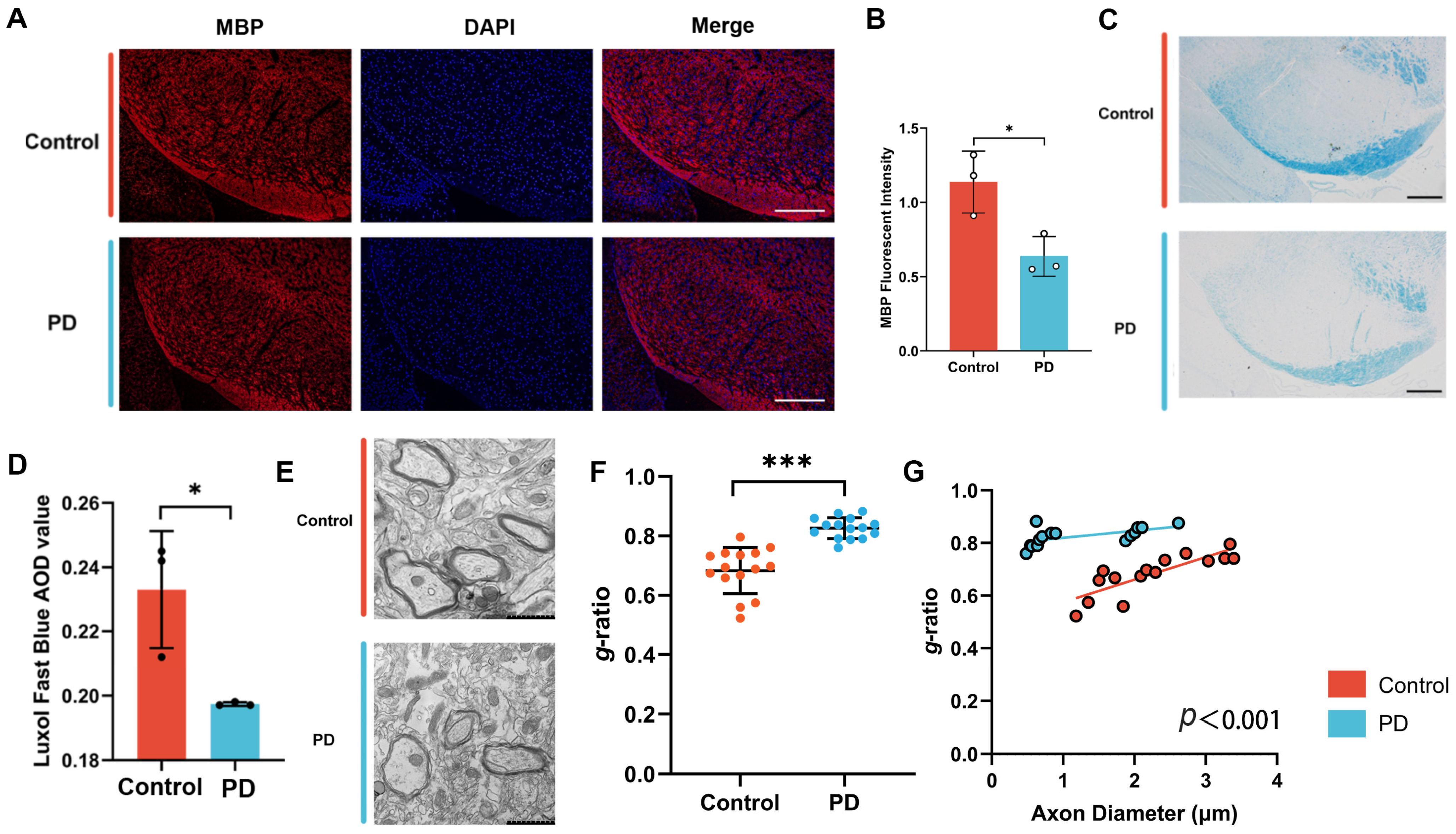
Paraffin sections (5 μm thick) were deparaffinized in xylene. Sections were then stained overnight at room temperature with Luxol Fast Blue (LFB, catalog number: G3242, Solarbio, Beijing, China). After staining, they were washed with 95% ethanol and distilled water. Differentiation was performed using a Fast Blue differentiation solution for 15 s, followed by a 30 s treatment with 70% ethanol until a clear distinction between gray and white matter was achieved. The sections were subsequently washed with distilled water, quickly dehydrated in 95% and 100% ethanol, cleared in xylene, and mounted. Slides were air-dried for one week prior to imaging. Images were captured using an OLYMPUS BX53 microscope (Olympusm, Tokyo, Japan). The average optical density (AOD) was measured using the ImageJ (v1.51) software.
2.8. Transmission Electron Microscopy
Mice were perfused with freshly prepared electron microscopy perfusion solution, ensuring that tissue collection was completed within one minute post-euthanasia. The extracted brain tissue was promptly placed in a low-temperature ice box. Electron microscopy fixative was uniformly applied to the tissue surface, and a 1 × 1 × 1 mm3 tissue block, essential for effective fixative penetration, was carefully excised with a blade. The excised samples were then stored overnight at 4 °C. The following day, the samples were dispatched to the Transmission Electron Microscopy Laboratory at Hebei Medical University’s Large Instrument Experiment Platform for further experimental procedures. Myelin sheath observations in the SN of the mice were conducted using a HT7800 transmission electron microscope (Hitachi, Tokyo, Japan).
2.9. Cell Culture and Differentiation
The MO3.13 human oligodendroglia cell line (catalog number: CL-0772, Wuhan Pricella Biotechnology Co., Ltd., Wuhan, China) was cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (catalog number: 11965092, Gibco, Shanghai, China) supplemented with 20% fetal bovine serum (FBS, catalog number: FB15015, Clerk, VA, USA) and 1% penicillin/streptomycin solution (catalog number: P1400, Solarbio, Beijing, China) in an H
2O-saturated 5% CO
2 atmosphere at 37 °C. For differentiation, once cells reached 70–80% confluency in poly-L-lysine-coated 24-well or 6-well plates, they were shifted to a serum-free differentiation medium containing 100 nM phorbol myristate acetate (PMA, catalog number: HY-18739, MedChemExpress, Shanghai, China). The medium was changed every 3 days, and cells were cultured for 7 days to induce differentiation [
17]. Following the induced differentiation period, cells were subjected to drug intervention, transfection, and subsequent protein and RNA collection.
The SH-SY5Y human neuroblastoma cell line was cultured in DMEM/F12 medium (catalog number: 11320033, Gibco, Shanghai, China) supplemented with 10% FBS and 1% penicillin/streptomycin solution. The cells were maintained in an H2O-saturated 5% CO2 atmosphere at 37 °C. SH-SY5Y cells were differentiated by treatment with 10 μM retinoic acid (RA; catalog number: HY-14649, MedChemExpress, Shanghai, China) in serum-free or low-serum medium for 3–5 days. Following differentiation, neuronally differentiated cells were co-cultured with differentiated MO3.13 cells and subjected to LFB staining. Both cell lines were recently authenticated by STR profiling and tested for mycoplasma contamination, with all results being negative.
2.10. CCK-8 Assay
Cell viability was evaluated using the CCK-8 assay (catalog number: K1018; APExBIO, Shanghai, China). Briefly, 5 × 103 cells/well were plated into 96-well plates and incubated for 24 h. Subsequently, cells were exposed to 1-methyl-4-phenylpyridinium iodide (MPP+) for various time intervals: 4, 6, 12, 24, and 48 h. After the treatment, 10 µL of CCK-8 reagent was added to each well, and cells were further incubated for 1 h. Absorbance (optical density) was recorded at 450 nm using a microplate reader. This experiment was performed in triplicate, with three wells per experimental condition. Data were analyzed using SPSS, and comparisons between treatment groups were conducted to identify significant differences, with a p-value < 0.05 considered statistically significant.
2.11. Transfection of Plasmids
The plasmids used in this study were constructed by GenScript. For plasmid extraction, Escherichia coli was cultured in LB liquid medium, and plasmids were extracted using a plasmid extraction kit from TIANGEN (catalog number: DP123, Beijing, China). The extraction process involved cell collection, resuspension, lysis, precipitation to remove impurities, washing, centrifugation, and elution, followed by plasmid concentration determination. For transfection, when the cells reached 60–70% confluency, plasmids and UltraFection 3.0 transfection reagent (catalog number: ELS300, SiZhengBai, Suzhou, China) were prepared and mixed and then incubated. The medium was not changed within 24 h after plasmid addition. qRT-PCR was performed 24 h post-transfection, and Western blot analysis was conducted 48 h post-transfection.
2.12. Transfection of siRNA
The siRNA constructs used in this study were provided by GenePharma. MO3.13 cells were passaged in 6-well plates and induced to differentiate before siRNA transfection. The CALNP™ RNAi in vitro transfection kit (catalog number: DN001-10, D-Nano Therapeutics Technology Co., Ltd., Beijing, China) was used for this purpose. A total of 5 µL of 20 µM siRNA solution was mixed with 40 µL of Solution A and 10 µL of Solution B, followed by the addition of 145 µL of cell culture medium, resulting in a total volume of 200 µL, which was then added to the 6-well plate containing 2 mL of the culture medium. The medium was not changed within 24 h after siRNA addition. qRT-PCR was performed 24 h post-transfection, and Western blot analysis was conducted 48 h post-transfection.
2.13. RT-qPCR
Total RNA was extracted using the Total RNApure Reagent Kit (ZP401, ZOMANBIO, Beijing, China), and 1 μg RNA was reverse-transcribed using HiScript III RT SuperMix (R323, Vazyme, Nanjing, China) under the following conditions: 42 °C (2 min), 37 °C (15 min), and 85 °C (5 s). RT-qPCR was performed with ChamQ Universal SYBR qPCR Master Mix (KT201, Vazyme, Nanjing, China) on a QuantStudio™ 6 Flex system (4484642, Applied Biosystems, Shanghai, China) under standardized cycling conditions: 95 °C (15 min), followed by 40 cycles of 95 °C (10 s), 60 °C (30 s), and 72 °C (20 s). GAPDH served as the endogenous control, and the relative mRNA expression was calculated using the 2
−ΔΔCt method. All reactions were performed in triplicate, with statistical analysis conducted using two-tailed
t-tests and one-way ANOVA. Primer sequences are provided in
Table S1.
2.14. Western Blot
For protein sample preparation, the cells or mouse SN tissue were homogenized or lysed using RIPA buffer (catalog number: R0020, Solarbio, Beijing, China), which was augmented with PMSF 100 mM (catalog number: P0100, Solarbio, Beijing, China). Following ultrasonication, the samples were incubated on ice for 1 h. Protein solutions were clarified by centrifugation at 12,000× g for 20 min at 4 °C, and the supernatant was collected. The protein concentration was then determined using a BCA protein assay reagent kit (catalog number: PC0020, Solarbio, Beijing, China) and measured with an Infinite F200 plate reader (TECAN, Switzerland). The protein samples were combined with a 5× loading buffer (catalog number: P1040, Solarbio, Beijing, China). After heating, 20 μg of protein was resolved on a 10% SDS polyacrylamide gel, which was then electrotransferred onto polyvinylidene fluoride membranes. The membranes underwent blocking in 5% milk dissolved in TBST (0.1% Tween-20) for one hour before incubation with the following primary antibodies: rabbit anti-STAT5B (1:2000, catalog number: CSB-PA022815LA01HU, CUSABIO, Wuhan, China), mouse anti-GAPDH (1:5000, catalog number: GB120025, Servicebio, Wuhan, China), rabbit anti-MBP (1:200, catalog number: 10458-1-AP, Proteintech, Wuhan, China), and rabbit anti-TH (1:2000, catalog number: A5079, ABclonal, Wuhan, China). Subsequently, membranes were incubated with HRP-conjugated AffiniPure goat anti-rabbit IgG (H+L) (1:5000, catalog number: BF03008, Biodragon, Suzhou, China) and HRP-conjugated AffiniPure goat anti-mouse IgG (H+L) (1:5000, catalog number: SA00001-1, Proteintech, Wuhan, China), along with an Enhanced/Super ECL Kit (1:1, catalog number: BF06053-100, Biodragon, Suzhou, China). For statistical analysis, either two-tailed t-tests (for two groups) or one-way ANOVA (for three or more groups) were applied.
2.15. Animal Behavioral Experiments
2.15.1. Rotarod Experiment
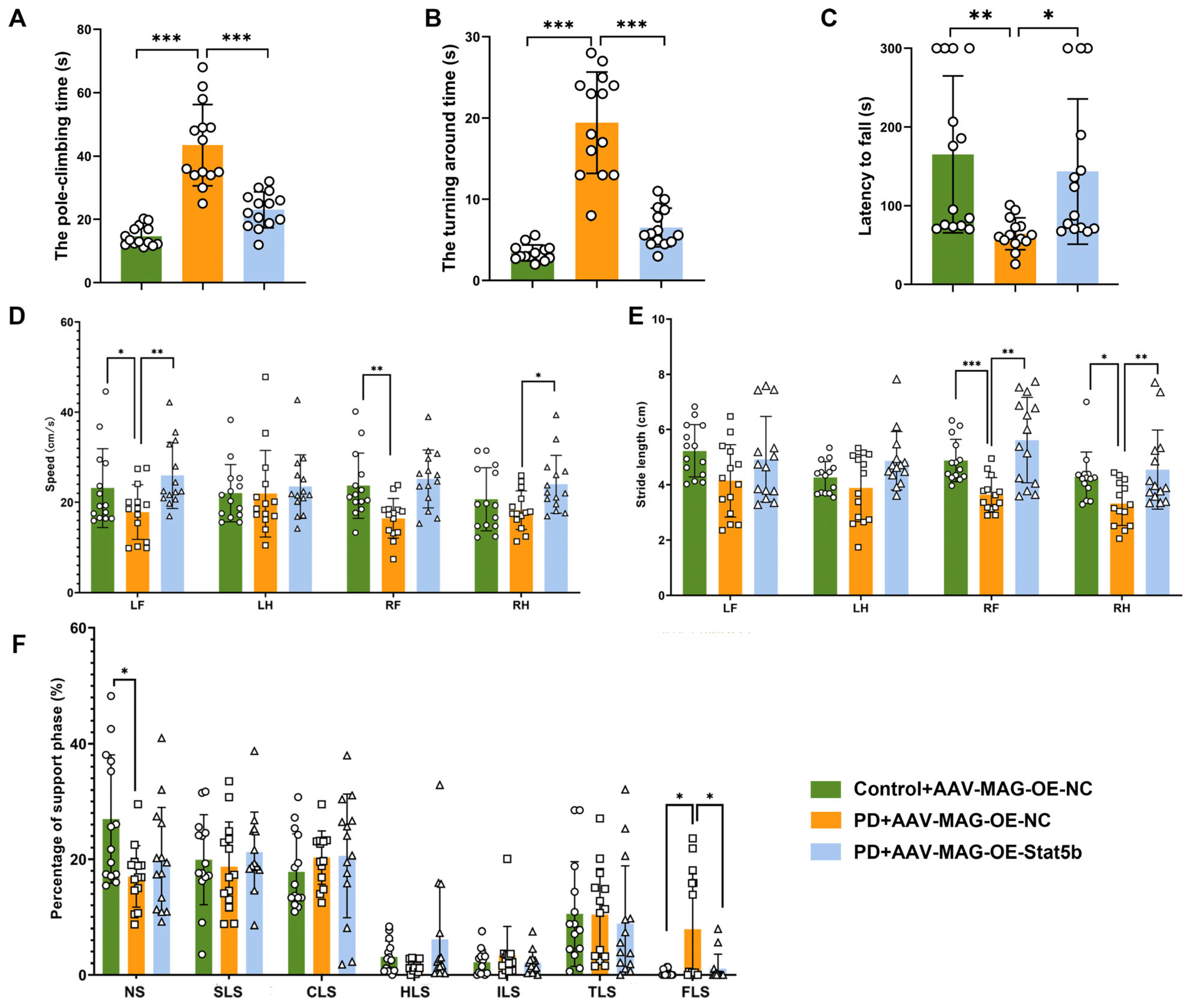
The rotarod assay was performed utilizing a mouse rotarod fatigue testing apparatus. The methodology was as follows. Each mouse was initially positioned onto the rotarod, which was set to an adjustable rotation speed of 25 revolutions per minute (rpm). A timer automatically documented the elapsed time from placement until the mouse dislodged from the rod (latency to fall). To ensure result reliability, a single measurement was conducted daily. By evaluating the duration of the mouse’s traversal on the rotarod, variations in motor performance were effectively analyzed.
2.15.2. Climbing Pole Experiment
Mice were placed head-up on a textured wooden sphere attached to a cylindrical pole within their home cage. Motor performance was quantified by measuring the turning around time and pole climbing time (T). The time taken for the mouse to navigate from the wooden sphere to the wooden pole while maintaining a downward head orientation was denoted as T1, measured with a stopwatch. Upon the mouse’s arrival at the lowest point of the pole, this time was noted as T2. The time taken to climb from the sphere to the pole base (T = T1 [initiation] − T2 [completion]) was recorded via stopwatch. Each mouse completed two trials, with the mean climbing duration used for analysis. The rough surface ensured a consistent grip during testing.
2.15.3. Gait Analysis
Gait assessment was conducted using an automated gait analysis system (Zhongshidi Technology Development Co., Ltd., Beijing, China). The mice were positioned at one terminus of the runway and permitted to traverse to the opposing end, where they could enter a dark enclosure. Each mouse underwent two housing sessions prior to the commencement of the formal experiment. The parameters measured encompassed the stride length, step velocity, and limb support time.
2.16. Bioinformatic Screening of Downstream Genes Using JASPAR and hTFtarget Databases
Potential downstream targets of STAT5B were identified through the systematic analysis of transcription factor binding sites (TFBSs) using the JASPAR and hTFtarget databases. Genes with significant associations (Q-value < 0.05) were retained after initial screening with an 80% relative score threshold. Four key parameters were normalized (0–1 range) and integrated into a composite score: the human TFBS count and maximum score (weight: 0.3 and 0.4, respectively); the mouse TFBS count and maximum score (combined weight: 0.3). The final prioritization considered both human data dominance and cross-species conservation evidence. This hierarchical approach not only prioritized human data but also integrated cross-species conservation evidence, thereby allowing for more comprehensive gene prioritization.
2.17. Luciferase Reporter Gene Assay
Here, 293T cells at ~60% confluence in 96-well plates were transfected in quintuplicate. After 24 h, luciferase activity was measured using the Dual-Luciferase® Reporter System (E1910, Promega, Beijing, China). Cells were PBS-washed, lysed with 50 μL passive lysis buffer/well (12 min dark agitation), and transferred to opaque plates. Firefly luciferase signals were quantified and then quenched with Stop & Glo® Reagent (50 μL/well) prior to Renilla luciferase measurement for normalization.
2.18. Actinomycin D Assay
MO3.13 cells were maintained in 6-well plates prior to treatment. Actinomycin D (ActD, catalog number: A4448, APExBIO, Shanghai, China) was added to each well at a final concentration of 10 μg/mL. Cells were then collected at designated time points following treatment over varying time intervals. Subsequently, total RNA was isolated, and the relative expression levels of STAT5B were evaluated using RT-qPCR.
2.19. MethylTarget Sequencing
The MethylTarget™ sequencing was performed by E-GENE Tech Co., Ltd., located in Shenzhen, China [
18]. The EZ DNA Methylation-Gold™ Kit (catalog number: DP215, TIANGEN Biotech, Beijing, China) was utilized to convert all unmethylated cytosines into uracils. Any samples that showed a bisulfite conversion rate of less than 98% were excluded from further analysis. Following the amplification, separation, and purification of the target CpG regions, a CpG island methylation assay was conducted using an Illumina Hiseq/Miseq 2000 (San Diego, CA, USA), according to the manufacturer’s instructions.
2.20. DNA Extraction and Bisulfite Conversion
Genomic DNA was extracted using the Biospin Genomic DNA Extraction Kit from Sangon Biotech (catalog number: B8251, Shanghai, China), according to the manufacturer’s instructions. For the bisulfite conversion process, 500 ng of the purified DNA was treated with the EZ DNA Methylation-Gold™ Kit from TIANGEN Biotech (catalog number: DP215, Beijing, China). The procedure involved DNA denaturation at 98 °C for 10 min, followed by bisulfite conversion at 64 °C for 2.5 h. After conversion, the DNA was purified using spin columns and then eluted in 20 μL of Tris-EDTA buffer (pH 8.0) and stored at –20 °C until it was needed for further analysis.
2.21. Methylation-Specific PCR Assay
Methylation-specific PCR (MSP) was conducted to assess the methylation status using the MSP Kit from TIANGEN Biotech (catalog number: EM101, Beijing, China), which contains primers specifically designed for bisulfite-converted sequences. The reaction mixture was prepared in a total volume of 20 μL, comprising less than 500 ng of template DNA, 1 μL each of the forward and reverse primers at a concentration of 10 μM, 1.6 μL of dNTPs at 2.5 mM, 1 unit of MSP DNA polymerase, 2 μL of 10× MSP buffer, and nuclease-free water to achieve the final volume. The thermocycling protocol began with an initial denaturation step at 95 °C for 5 min, followed by 35 cycles of amplification at 94 °C for 20 s, 60 °C for 30 s, and 72 °C for 20 s. A final extension step was performed at 72 °C for 5 min. The PCR products (approximately 400 bp) were subsequently analyzed by 2% agarose gel electrophoresis and visualized under UV light.
2.22. Statistical Analysis
Data are presented as means ± standard error of the mean (SEM). To evaluate statistical differences between the two groups, Student’s t-test was utilized. For analyses involving multiple groups, one-way ANOVA was performed, followed by either the least significant difference (LSD) test or Dunnett’s T3 post hoc test. All statistical evaluations were conducted using SPSS version 26. Data visualization was executed with GraphPad Prism version 8.0.2. Correlations were assessed using Pearson correlation analysis, with a p-value of less than 0.05 considered statistically significant.
4. Discussion
Our study significantly strengthens the understanding of oligodendrocytes’ involvement in PD pathogenesis. We identified a novel epigenetic pathway where DNMT3A-mediated hypermethylation represses STAT5B expression in oligodendrocytes, leading to the subsequent downregulation of MBP and myelin injury, ultimately exacerbating dopaminergic neuronal degeneration.
Oligodendrocytes are not only responsible for the formation and maintenance of the myelin sheath but are also actively involved in neuroprotective and repair processes. However, conventional research methods often fall short in fully capturing the heterogeneity of oligodendrocytes and their specific functions in PD. The advent of scRNA-seq has revolutionized this field, enabling the in-depth exploration of oligodendrocytes’ diversity and their role in PD at the single-cell level [
22]. Our snRNA-seq analysis of the MPTP-induced mouse model revealed a decreased proportion of oligodendrocytes and the pervasive downregulation of myelin-related genes in the SN. Monocle2 pseudotime analysis further elucidated the dynamic transitions between oligodendrocyte subpopulations, revealing the accumulation of PD group cells at terminal differentiation states characterized by progressive myelin gene silencing. Furthermore, a scRNA-seq atlas of the human SN revealed a significant association between PD risk and oligodendrocyte-specific gene expression [
23]. Another scRNA-seq analysis demonstrated the dysregulation of immune regulation and cytokine signaling pathways in oligodendrocytes and oligodendrocyte precursor cells (OPCs) from PD patients, consistent with the neuroinflammatory processes observed in PD [
24]. Recent studies further support these findings, demonstrating that GPR37 upregulation and PSAP secretion in oligodendrocytes contribute to IL-6 secretion, neuroinflammation, and dopaminergic neuron degeneration in an MPTP-induced mouse model [
25]. Furthermore, reports of reduced oligodendrocyte densities in the SN and frontal cortex in PD patients, coupled with a decrease in oligodendrocyte precursor cells in the PD brain [
26], collectively suggest impaired myelin maintenance and repair mechanisms in individuals with PD [
27]. STAT5B plays a key role in regulating the activity and survival of dopaminergic neurons [
28].
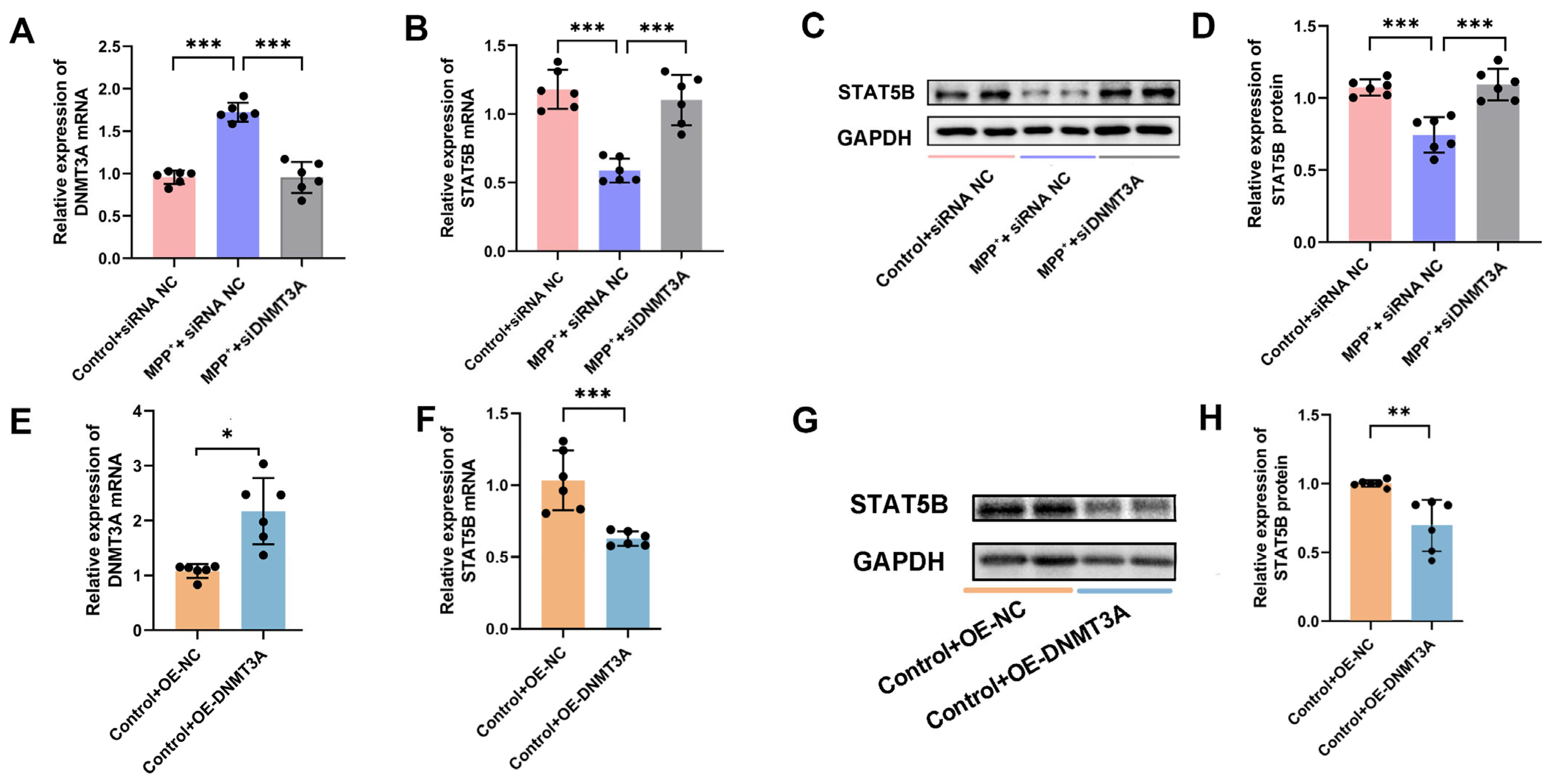
STAT5B knockout mice exhibit significantly reduced brain dopamine levels, increased dopaminergic neuron mortality, and abnormal behaviors, including decreased locomotor activity and anxiety. The overexpression of
STAT5B enhances the association between dopaminergic neurons and presynaptic neurons, thereby increasing dopaminergic neuron activity and dopamine release [
15]. In PD mouse models, activated STAT5B has been shown to inhibit mitochondrial fission and attenuate degenerative damage in dopaminergic neurons [
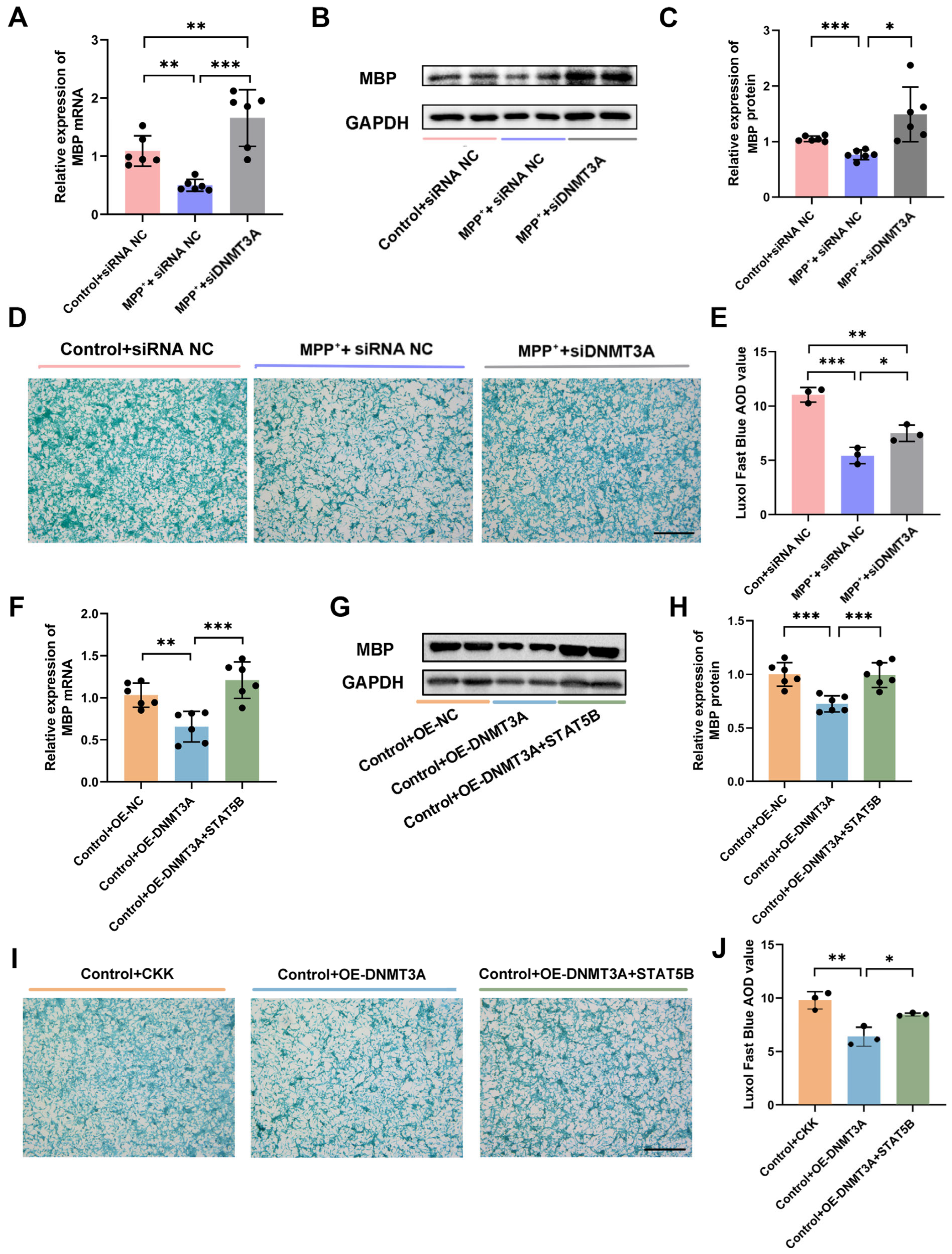
29]. Our results demonstrated that
STAT5B overexpression significantly improved myelination damage, protected dopaminergic neurons, and ameliorated motor impairments in the MPTP-induced mouse model. The consistent findings across both the in vivo and in vitro experiments further validate the crucial role of STAT5B in mitigating myelin injury and pathological progression in oligodendrocytes. While the MPTP model effectively recapitulates key dopaminergic neurodegeneration and motor dysfunction aspects that are central to PD pathogenesis, it is important to acknowledge its potential for direct neurotoxic effects on various cell types, including oligodendrocytes. Nevertheless, the increasing recognition of myelin dysfunction and oligodendrocyte alterations in human PD brain tissue and other non-MPTP PD models (e.g., alpha-synucleinopathy models) [
30,
31] suggests that the observed STAT5B-mediated myelin impairment in our study may represent a relevant pathological feature in a broader spectrum of PD etiologies.
MBP is a critical structural component of the central nervous system myelin sheath [
32], predominantly expressed in mature oligodendrocytes and essential for myelin formation and maintenance [
33,
34]. Previous studies have demonstrated that apotransferrin can regulate
MBP expression in oligodendrocytes through various signaling pathways and TFs [
35]. Notably, Sox10 is instrumental in myelination, interacting with Sp1 to promote
MBP gene transcription [
36]. Additionally, p27Kip1 has been shown to enhance
MBP expression by stabilizing the Sp1 protein and facilitating its binding to the
MBP promoter [
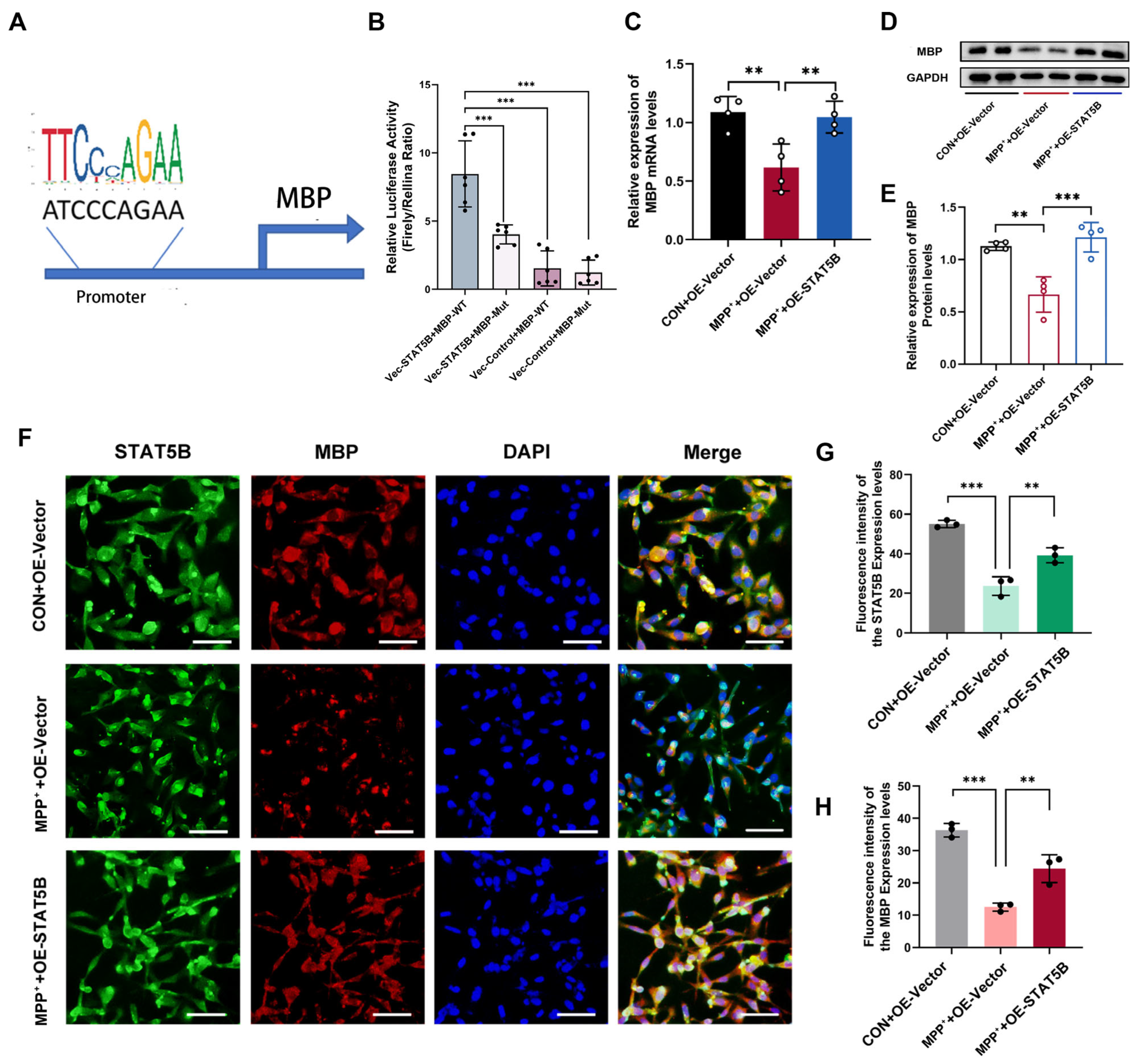
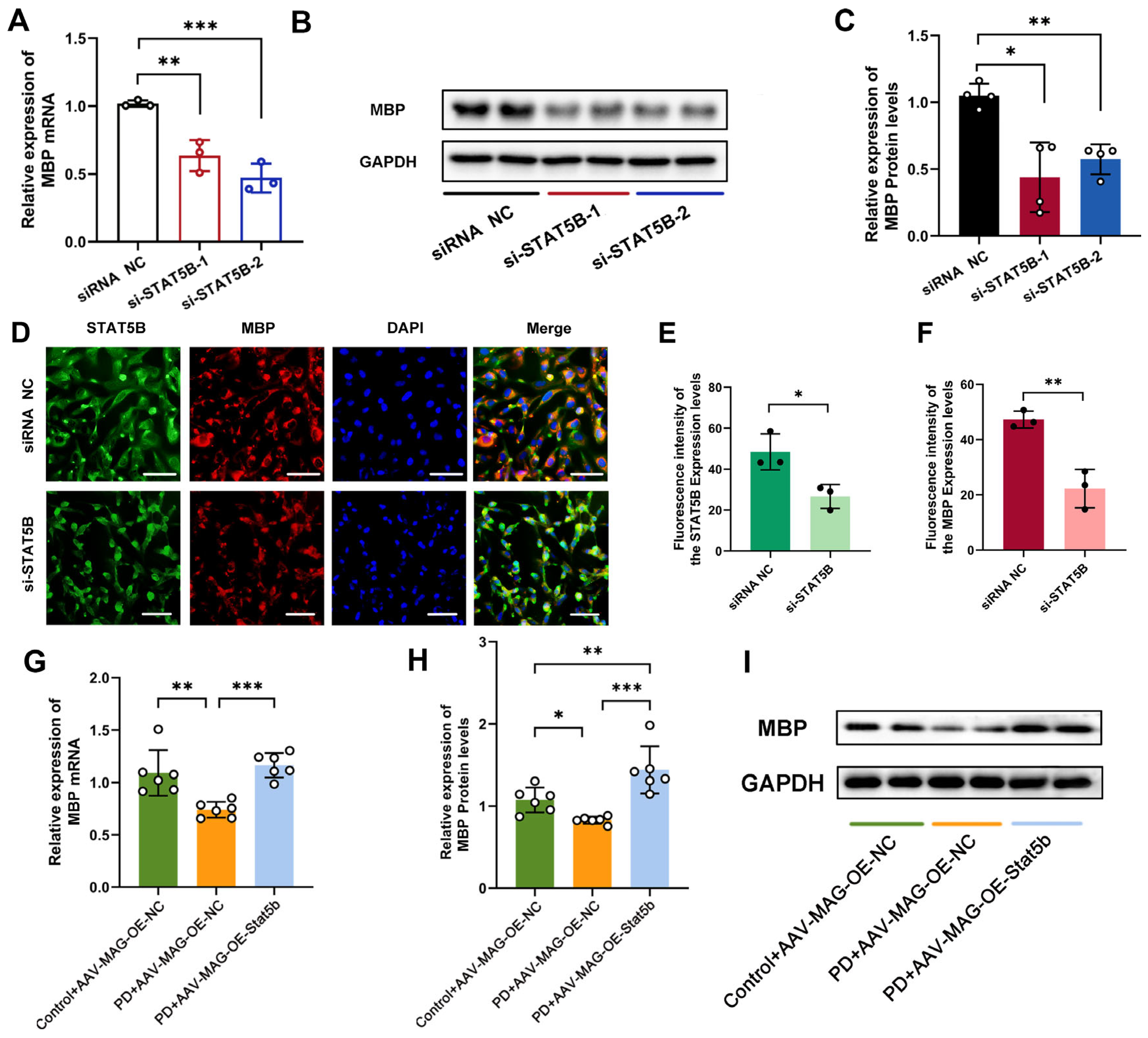
37]. Our study extends these findings by employing a luciferase reporter gene assay to identify STAT5B as a key transcriptional regulator of
MBP. Furthermore, we demonstrate that
STAT5B directly binds to the
MBP promoter, thereby activating its expression and profoundly influencing myelin formation and maintenance.
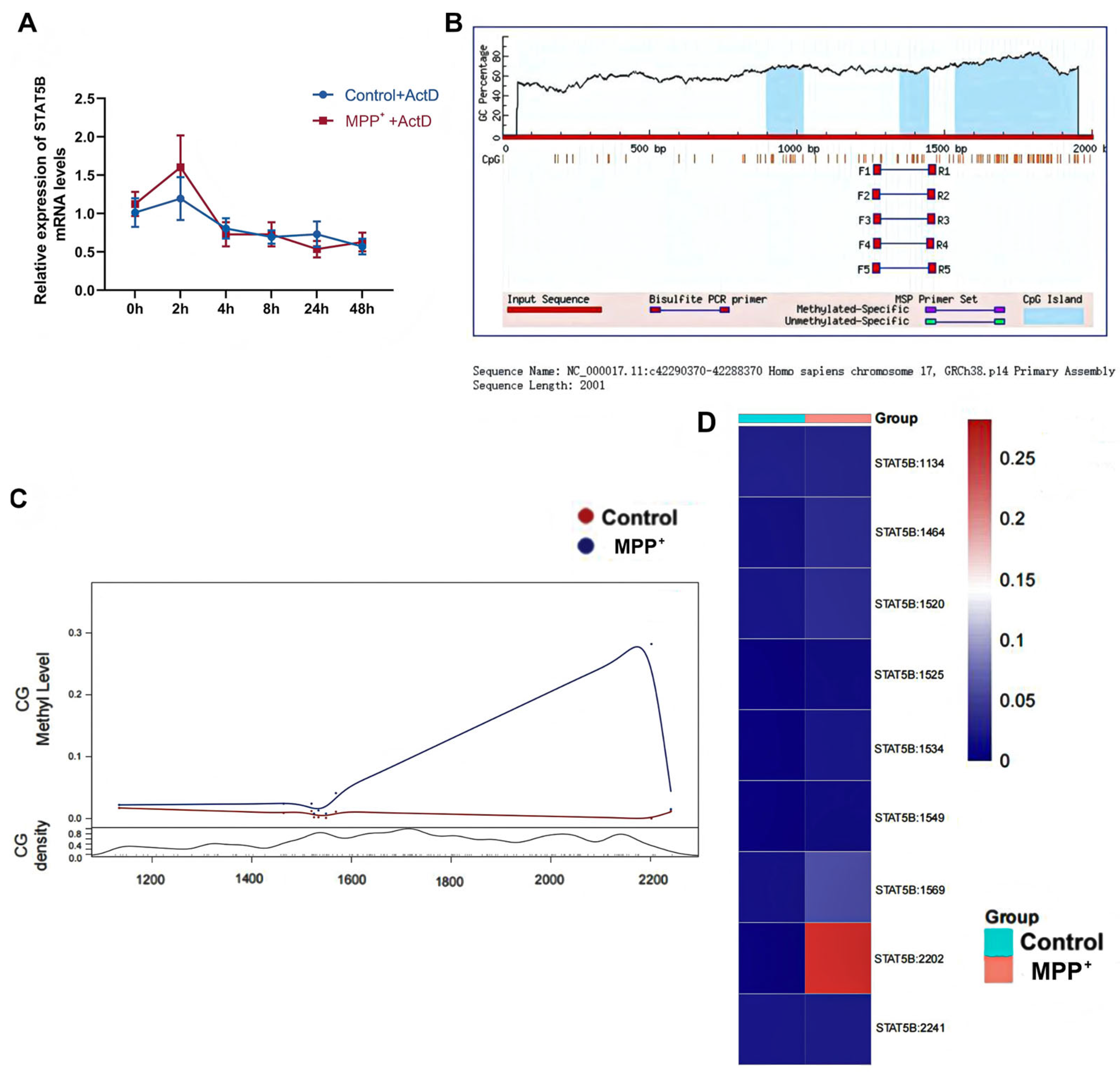
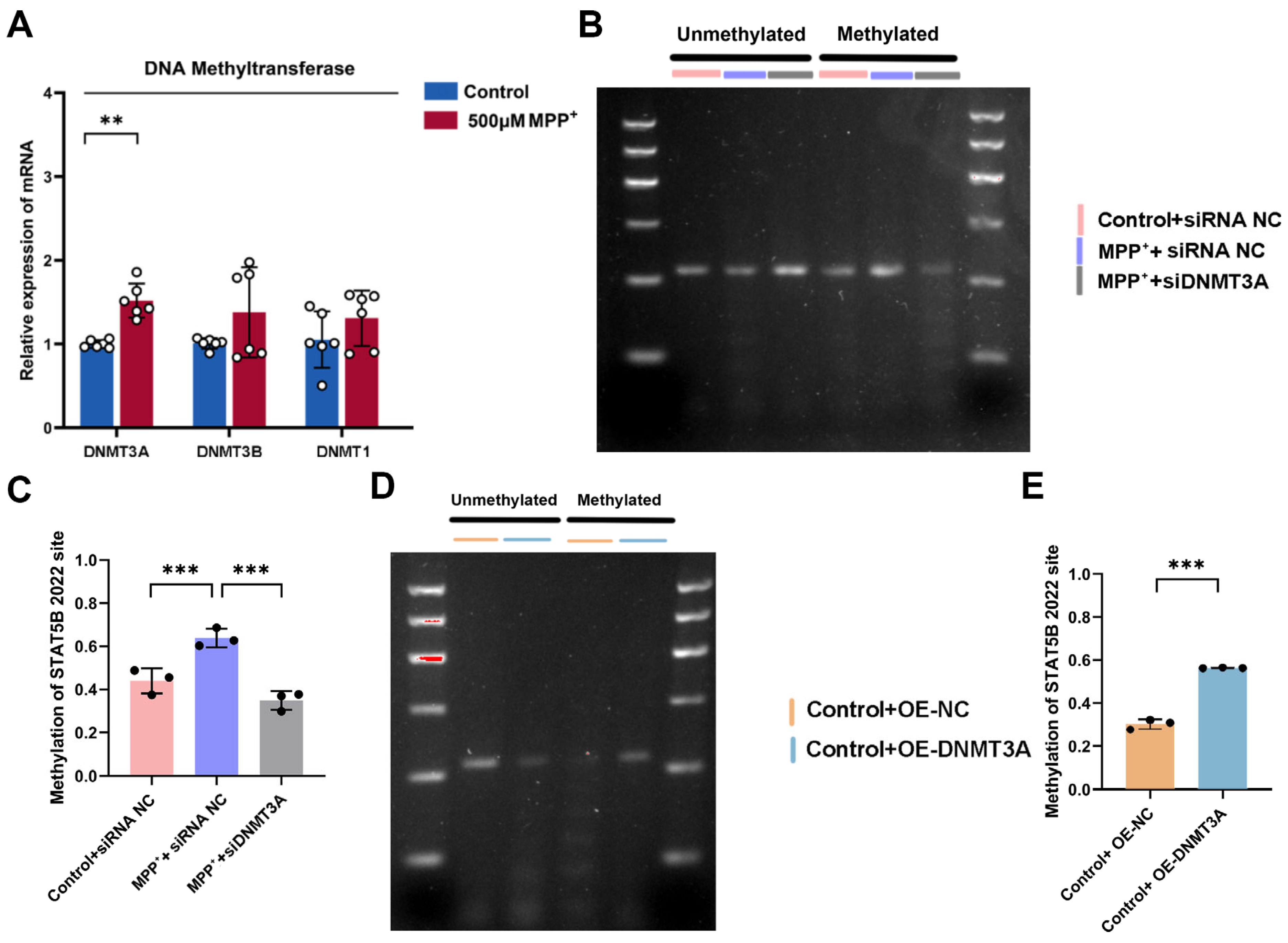
To investigate the upstream mechanisms contributing to reduced
STAT5B transcription in oligodendrocytes, we utilized ActD as a transcriptional inhibitor. ActD reduces RNA synthesis by inhibiting RNA polymerase, enabling us to assess
STAT5B mRNA stability independently of newly synthesized transcripts. Our results indicate that the MPP
+-induced decrease in
STAT5B mRNA levels is not primarily due to accelerated mRNA degradation but rather strongly suggests a reduced transcriptional rate. DNA methylation, a key epigenetic mechanism in regulating transcriptional activity, typically suppresses gene expression by methylating promoter regions, thereby hindering transcription factor binding or recruiting repressive complexes [
38]. Bioinformatics analysis predicted dense CpG islands within the
STAT5B promoter, while concurrent qRT-PCR revealed significantly upregulated
DNMT3A expression following MPP
+ treatment. This strongly suggests that DNMT3A-mediated DNA methylation contributes to
STAT5B transcriptional repression, a notion corroborated by studies implicating DNMT3A in PD pathogenesis, such as its role in modulating SNCA expression through targeted methylation [
39,
40]. Furthermore, the principle of DNMT3A epigenetically silencing crucial glial genes and impairing myelination is not without precedent; for instance, in multiple sclerosis, the hypermethylation of myelination-related genes, including
MBP, by DNMT3A-implicated mechanisms contributes to oligodendrocyte dysfunction [
41]. Our study, therefore, identifies for the first time the DNMT3A-
STAT5B-
MBP axis in oligodendrocytes as a direct regulatory pathway, providing compelling evidence for DNMT3A-driven glial epigenetic dysregulation and subsequent myelin injury in PD pathogenesis.
In summary, our comprehensive findings elucidate novel molecular mechanisms underlying oligodendrocyte alterations and myelin damage in the SN of the MPTP-induced mouse model, significantly contributing to the understanding of PD pathogenesis and providing specific gene expression signatures. Our future studies will focus on validating these findings in human PD patient samples and patient-derived cell models. A deeper understanding of oligodendrocyte epigenetic dysregulation during PD progression holds immense promise in revealing novel therapeutic targets, thereby establishing a foundation for the development neuroprotective strategies specifically focused on myelin repair in the treatment of PD.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}