.png)
CRISPR/Cas-Based Ex Vivo Gene Therapy and Lysosomal Storage Disorders: A Perspective Beyond Cas9



Abstract
1. Introduction
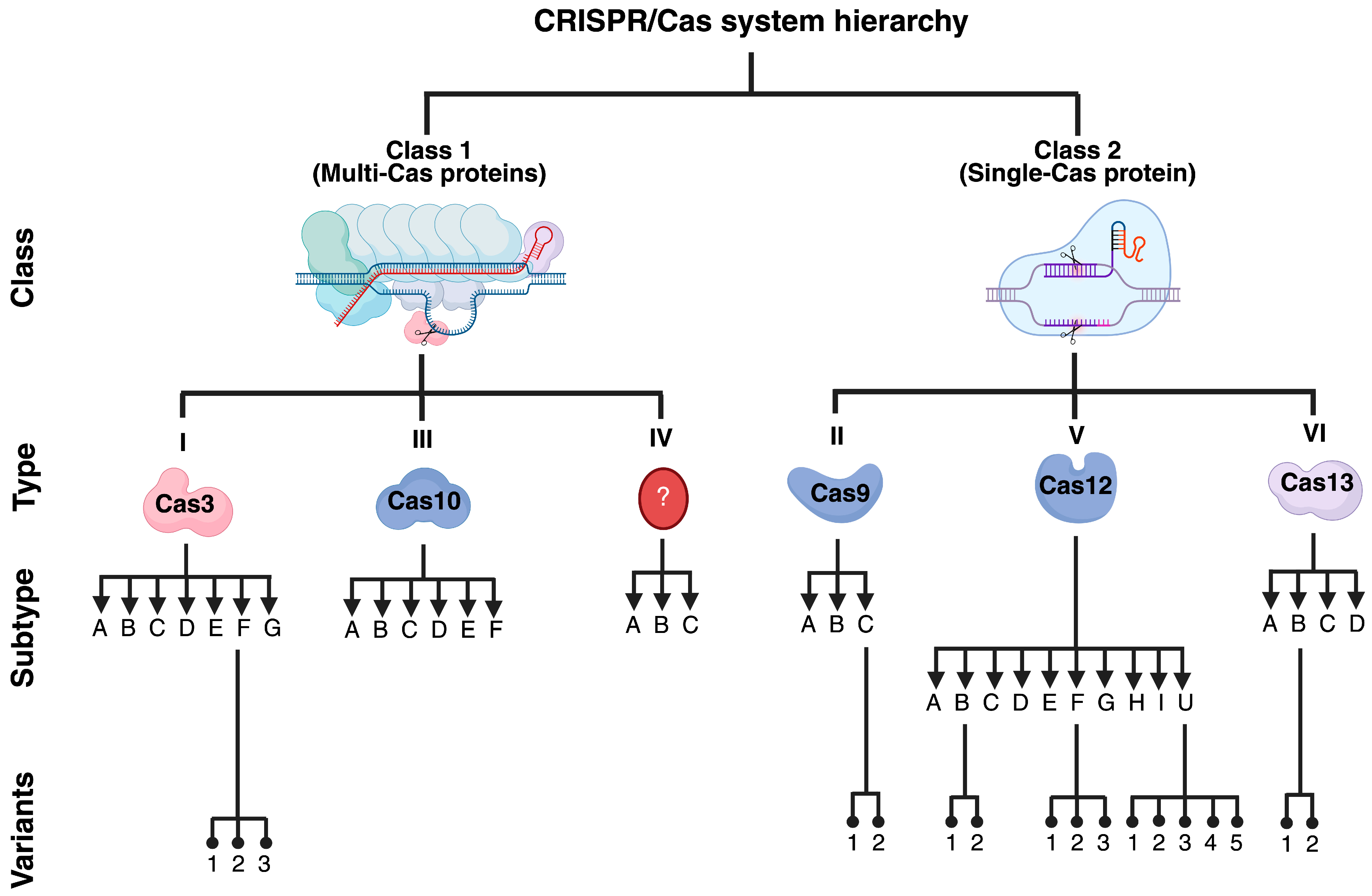
2. The CRISPR/Cas Systems
2.1. DNA-Targeting Cas Enzymes
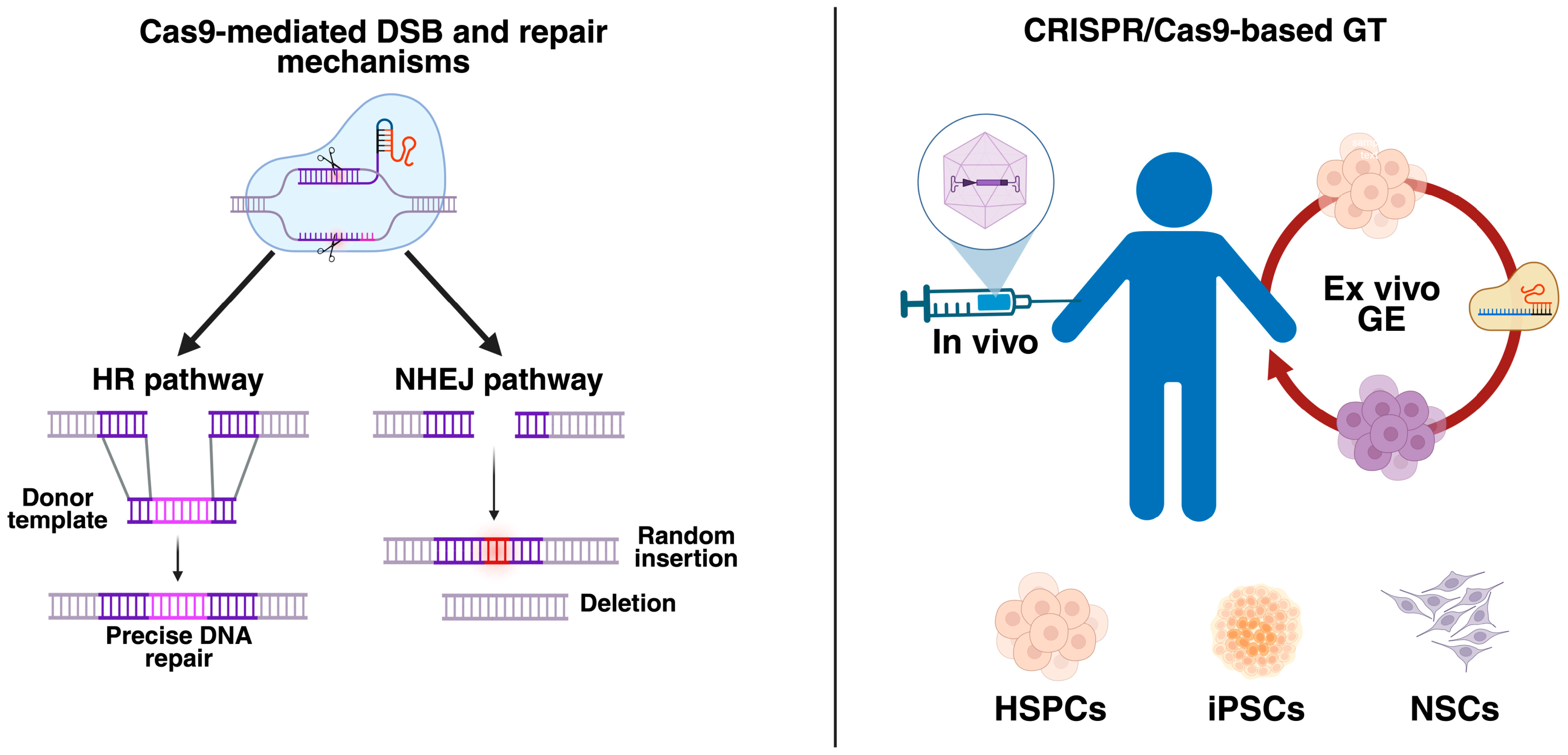
2.1.1. Cas9
2.1.2. Cas3
2.1.3. Cas12
2.1.4. Cas14
2.2. RNA-Targeting Cas Enzymes
2.2.1. Cas7-11
2.2.2. Cas13
2.3. CRISPR/Cas-Derived Alternatives
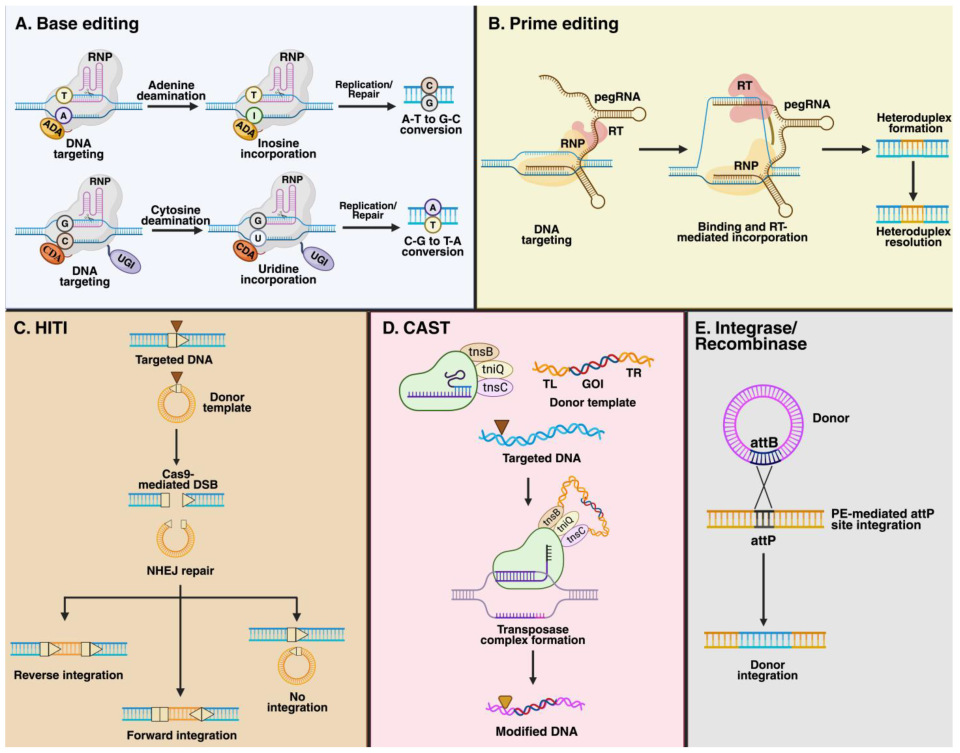
2.3.1. Base Editing
2.3.2. Prime Editing (PE)
2.3.3. Homology-Independent Targeted Integration (HITI)
3. CRISPR/Cas9: Ex Vivo Approaches in LSDs
3.1. Mucopolysaccharidosis (MPS) I
3.2. Mucopolysaccharidosis (MPS) IVA
3.3. Gaucher
3.4. Krabbe
3.5. Fabry
4. Beyond Cas9 Enzymes and HR: An Overview in Non-LSD Models
4.1. CAR-T Cells: Implementing Cas12a
4.2. Hematological Disorders: Base and Prime Editing
4.3. HITI: Advancing in Proof-of-Concept Assays
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BE | Base editing |
| BM | Bone marrow |
| DSB | Double-strand break |
| GAG | Glycosaminoglycan |
| GE | Genome editing |
| GT | Gene therapy |
| HR | Homologous recombination |
| HSCT | Hematopoietic stem cell transplantation |
| HSPCs | Hematopoietic stem progenitor cells |
| iPSCs | Induced pluripotent stem cells |
| NHEJ | Non-homologous end joining |
| PE | Prime editing |
| WT | Wild type |
References
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal storage diseases: Current therapies and future alternatives. J. Mol. Med. 2020, 98, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pereira, C.; Millán-Tejado, B.S.; Gallardo-Gómez, M.; Pérez-Márquez, T.; Alves-Villar, M.; Melcón-Crespo, C.; Fernández-Martín, J.; Ortolano, S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules 2021, 11, 1775. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Jaramillo, R.G.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022, 13, 975803. [Google Scholar] [CrossRef] [PubMed]
- Pupo, A.; Fernández, A.; Low, S.H.; François, A.; Suárez-Amarán, L.; Samulski, R.J. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 2022, 30, 3515–3541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Anselmo, A.C.; Mitragotri, S. Viral vector-based gene therapies in the clinic. Bioeng. Transl. Med. 2022, 7, e10258. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Herreno-Pachón, A.M.; Benincore-Flórez, E.; Karunathilaka, A.; Tomatsu, S. Current Strategies for Increasing Knock-In Efficiency in CRISPR/Cas9-Based Approaches. Int. J. Mol. Sci. 2024, 25, 2456. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, J.; Yun, S.; Xue, C.; Yao, Y.; Rao, S. Recent advances in CRISPR-Cas9-based genome insertion technologies. Mol. Ther. Nucleic Acids 2024, 35, 102138. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Celik, B.; Fnu, N.; Khan, S.; Tomatsu, S.; Alméciga-Díaz, C.J. Iron oxide-coupled CRISPR-nCas9-based genome editing assessment in mucopolysaccharidosis IVA mice. Mol. Ther. Methods Clin. Dev. 2023, 31, 101153. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; Gonzalez, E.A.; Tavares, A.M.V.; Seolin, B.G.; De Elias, L., Sr.; Vera, L.N.P.; Kubaski, F.; Poletto, E.; Giugliani, R.; Teixeira, H.F.; et al. Neonatal nonviral gene editing with the CRISPR/Cas9 system improves some cardiovascular, respiratory, and bone disease features of the mucopolysaccharidosis I phenotype in mice. Gene Ther. 2020, 27, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; Poletto, É.; Pasqualim, G.; Tavares, A.M.V.; Meyer, F.S.; Gonzalez, E.A.; Giugliani, R.; Matte, U.; Teixeira, H.F.; Baldo, G. In vivo genome editing of mucopolysaccharidosis I mice using the CRISPR/Cas9 system. J. Control. Release 2018, 288, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Poletto, E.; Silva, A.O.; Weinlich, R.; Martin, P.K.M.; Torres, D.C.; Giugliani, R.; Baldo, G. Ex vivo gene therapy for lysosomal storage disorders: Future perspectives. Expert. Opin. Biol. Ther. 2023, 23, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Pará, C.; Bose, P.; Pshezhetsky, A.V. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J. Clin. Med. 2020, 9, 616. [Google Scholar] [CrossRef] [PubMed]
- Ellison, S.; Parker, H.; Bigger, B. Advances in therapies for neurological lysosomal storage disorders. J. Inherit. Metab. Dis. 2023, 46, 874–905. [Google Scholar] [CrossRef] [PubMed]
- Capotondo, A.; Milazzo, R.; Politi, L.S.; Quattrini, A.; Palini, A.; Plati, T.; Merella, S.; Nonis, A.; di Serio, C.; Montini, E.; et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2012, 109, 15018–15023. [Google Scholar] [CrossRef] [PubMed]
- Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef] [PubMed]
- Poletto, E.; Colella, P.; Vera, L.N.P.; Khan, S.; Tomatsu, S.; Baldo, G.; Gomez-Ospina, N. Improved engraftment and therapeutic efficacy by human genome-edited hematopoietic stem cells with Busulfan-based myeloablation. Mol. Ther. Methods Clin. Dev. 2022, 25, 392–409. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hemato-poietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transpl. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, K.; Uygun, V.; Hismi, B.O.; Celen, S.; Ozturkmen, S.; Zhumatayev, S.; Daloglu, H.; Karasu, G.; Yesilipek, A. Hematopoietic stem cell transplantation in children with mucopolysaccharidosis IVA: Single center experience. Bone Marrow Transplant. 2024, 60, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Rossini, L.; Durante, C.; Marzollo, A.; Biffi, A. New Indications for Hematopoietic Stem Cell Gene Therapy in Lysosomal Storage Disorders. Front. Oncol. 2022, 12, 885639. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Holler, E.; Sandmaier, B.M.; Huang, H.; Mohty, M. Acute graft-versus-host disease. Nat. Rev. Dis. Primers 2023, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Fnu, N.; Benincore-Flórez, E.; Herreño-Pachón, A.M.; Echeverri-Peña, O.Y.; Alméciga-Díaz, C.J.; Tomatsu, S. The landscape of CRISPR/Cas9 for inborn errors of metabolism. Mol. Genet. Metab. 2022, 138, 106968. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endo-nuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Hryhorowicz, M.; Lipiński, D.; Zeyland, J. Evolution of CRISPR/Cas Systems for Precise Genome Editing. Int. J. Mol. Sci. 2023, 24, 14233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Nam, K.H.; Ding, F.; Lee, H.; Wu, L.; Xiao, Y.; Farchione, M.D., Jr.; Zhou, S.; Rajashankar, K.; Kurinov, I.; et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014, 21, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Lee, S.Y.; Ha, H.J.; Park, H.H. Structural basis of Cas3 activation in type I-C CRISPR-Cas system. Nucleic Acids Res. 2024, 52, 10563–10574. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, H.; Han, Y.; Song, Y.; Wei, M.; Fang, M.; Sun, Y. CRISPR/Cas9 Gene Editing System Can Alter Gene Expression and Induce DNA Damage Accumulation. Genes 2023, 14, 806. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed]
- Csörgő, B.; León, L.M.; Chau-Ly, I.J.; Vasquez-Rifo, A.; Berry, J.D.; Mahendra, C.; Crawford, E.D.; Lewis, J.D.; Bondy-Denomy, J. A compact Cascade–Cas3 system for targeted genome engineering. Nat. Methods 2020, 17, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Morisaka, H.; Yoshimi, K.; Okuzaki, Y.; Gee, P.; Kunihiro, Y.; Sonpho, E.; Xu, H.; Sasakawa, N.; Naito, Y.; Nakada, S.; et al. CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 2019, 10, 5302. [Google Scholar] [CrossRef] [PubMed]
- Vaillend, C.; Aoki, Y.; Mercuri, E.; Hendriksen, J.; Tetorou, K.; Goyenvalle, A.; Muntoni, F. Duchenne muscular dystrophy: Recent insights in brain related comorbidities. Nat. Commun. 2025, 16, 1298. [Google Scholar] [CrossRef] [PubMed]
- Meliawati, M.; Schilling, C.; Schmid, J. Recent advances of Cas12a applications in bacteria. Appl. Microbiol. Biotechnol. 2021, 105, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, P.; Shi, Y.; Zhang, C.; Tong, X.; Fan, H.; Zhou, X.; Zhang, Y.; Yin, H. Characterization of a thermostable Cas12a ortholog. Cell Insight 2023, 2, 100126. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Wang, X.; Wang, Y.; Sun, Y.; Su, Y.; Lv, X.; Song, X. Advances in Cas12a-Based Amplification-Free Nucleic Acid Detection. CRISPR J. 2023, 6, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shen, X.; Chen, G.; Zhang, W.; Tan, B. Application and development of CRISPR-Cas12a methods for the molecular diagnosis of cancer: A review. Anal. Chim. Acta 2024, 1341, 343603. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Mention, K.; Cavusoglu-Doran, K.; Sanz, D.J.; Bacalhau, M.; Lopes-Pacheco, M.; Harrison, P.T.; Farinha, C.M. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282X-CFTR mutation. J. Cyst. Fibros. 2022, 21, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, S.; Lin, S.; Xing, W.; Yang, Y.; Zhu, F.; Su, D.; Chen, C.; Liu, J.-J.G. Cas12e orthologs evolve variable structural elements to facilitate dsDNA cleavage. Nat. Commun. 2024, 15, 10727. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Adiego-Pérez, B.; van der Oost, J. Biology and applications of CRISPR–Cas12 and transposon-associated homologs. Nat. Biotechnol. 2024, 42, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Li, Z.; Wang, S.; Han, R.; Chang, L. Structural basis for substrate recognition and cleavage by the dimerization-dependent CRISPR–Cas12f nuclease. Nucleic Acids Res. 2021, 49, 4120–4128. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.N.; Nakagawa, R.; Okazaki, S.; Hirano, H.; Kobayashi, K.; Kusakizako, T.; Nishizawa, T.; Yamashita, K.; Nishimasu, H.; Nureki, O. Structure of the miniature type V-F CRISPR-Cas effector enzyme. Mol. Cell 2021, 81, 558–570.e3. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Cai, B.; Tian, Y.; Liu, X.; Liang, C.; Gao, Q.; Li, B.; Ding, Y.; Li, R.; Zhou, Q.; et al. Therapeutic In Vivo Gene Editing Achieved by a Hypercompact CRISPR-Cas12f1 System Delivered with All-in-One Adeno-Associated Virus. Adv. Sci. 2024, 11, e2308095. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, X.; He, Q.; Wang, X.; Tang, J.; Wang, T.; Zhang, Y.; Yu, F.; Zhang, S.; Liu, Z.; et al. Structural basis for the activity of the type VII CRISPR–Cas system. Nature 2024, 633, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, R.; Sohail, M.; Kong, X.; Zhang, X.; Fu, N.; Li, B. CRISPR/Cas14 provides a promising platform in facile and versatile aptasensing with improved sensitivity. Talanta 2023, 254, 124120. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.F. Cas14: Big Advances from Small CRISPR Proteins. Biochemistry 2019, 58, 1024–1025. [Google Scholar] [CrossRef] [PubMed]
- Goswami, H.N.; Rai, J.; Das, A.; Li, H.; Institute of Molecular Biophysics, Florida State University, United States; Department of Chemistry and Biochemistry, Florida State University, United States. Molecular mechanism of active Cas7-11 in processing CRISPR RNA and interfering target RNA. eLife 2022, 11, 81678. [Google Scholar] [CrossRef] [PubMed]
- Özcan, A.; Krajeski, R.; Ioannidi, E.; Lee, B.; Gardner, A.; Makarova, K.S.; Koonin, E.V.; Abudayyeh, O.O.; Gootenberg, J.S. Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 2021, 597, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Li, H.; Brogan, D.J.; Wang, T.; Akbari, O.S.; A Komives, E. CRISPR RNA binding drives structural ordering that primes Cas7-11 for target cleavage. Nucleic Acids Res. 2025, 53, gkaf271. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, I.; Hernández-Huertas, L.; Nahón-Cano, D.; Martínez-García, P.M.; Treichel, A.J.; Gómez-Marin, C.; Tomás-Gallardo, L.; Pescador, G.d.S.; Kushawah, G.; Egidy, R.; et al. Enhanced RNA-targeting CRISPR-Cas technology in zebrafish. Nat. Commun. 2025, 16, 2591. [Google Scholar] [CrossRef] [PubMed]
- Yoon, P.H.; Zhang, Z.; Loi, K.J.; Adler, B.A.; Lahiri, A.; Vohra, K.; Shi, H.; Rabelo, D.B.; Trinidad, M.; Boger, R.S.; et al. Struc-ture-guided discovery of ancestral CRISPR-Cas13 ribonucleases. Science 2024, 385, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Patel, D.J. Structures, mechanisms and applications of RNA-centric CRISPR-Cas13. Nat. Chem. Biol. 2024, 20, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Hsiao, Y.-W.; Wong, V.H.Y.; Aubin, D.; Wang, J.-H.; Lisowski, L.; Rakoczy, E.P.; Li, F.; Alarcon-Martinez, L.; Gonzalez-Cordero, A.; et al. Characterization of RNA editing and gene therapy with a compact CRISPR-Cas13 in the retina. Proc. Natl. Acad. Sci. USA 2024, 121, e2408345121. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Belmonte, J.C.I. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2017, 63, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, G.; Johnson, W.A.; Qu, Y.; Yin, D.; Ramkissoon, N.; Xiang, H.; Cong, L. Long sequence insertion via CRISPR/Cas gene-editing with transposase, recombinase, and integrase. Curr. Opin. Biomed. Eng. 2023, 28, 100491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, J.; Wu, Y.; Xu, C.; Xu, P. Base Editors-Mediated Gene Therapy in Hematopoietic Stem Cells for Hematologic Diseases. Stem Cell Rev. Rep. 2024, 20, 1387–1405. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chen, F.; Wang, K.; Lai, L. Base editors: Development and applications in biomedicine. Front. Med. 2023, 17, 359–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Lian, M.; Ma, B.; Gou, S.; Luo, X.; Yang, K.; Shi, H.; Xie, J.; Ge, W.; Ouyang, Z.; et al. Multiplexed base editing through Cas12a variant-mediated cytosine and adenine base editors. Commun. Biol. 2022, 5, 1163. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; Bratovič, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Guo, D.; Wang, D.; Zhou, J.; Chen, Q.; Lai, J. Prime editing: A gene precision editing tool from inception to present. FASEB J. 2024, 38, e70148. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Gao, X.D.; Podracky, C.J.; Nelson, A.T.; Koblan, L.W.; Raguram, A.; Levy, J.M.; Mercer, J.A.M.; Liu, D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022, 40, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Doman, J.L.; Pandey, S.; Neugebauer, M.E.; An, M.; Davis, J.R.; Randolph, P.B.; McElroy, A.; Gao, X.D.; Raguram, A.; Richter, M.F.; et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell 2023, 186, 3983–4002.e26. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; He, Z.; Zhao, K.T.; Zhu, H.; Hu, J.; Liu, G.; Gao, Q.; Liu, M.; Zhang, R.; Qiu, J.-L.; et al. Prime editing using CRISPR-Cas12a and circular RNAs in human cells. Nat. Biotechnol. 2024, 42, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Jia, R.; Zhao, X.; Zhang, F.; Chen, S.; Yu, S.; Liu, X.; Dou, H.; Feng, X.; Zhang, J.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar]
- Miki, T.; Vazquez, L.; Yanuaria, L.; Lopez, O.; Garcia, I.M.; Ohashi, K.; Rodriguez, N.S. Induced Pluripotent Stem Cell Derivation and Ex Vivo Gene Correction Using a Mucopolysaccharidosis Type 1 Disease Mouse Model. Stem Cells Int. 2019, 2019, 6978303. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef] [PubMed]
- Herreño-Pachón, A.M.; Leal, A.F.; Khan, S.; Alméciga-Díaz, C.J.; Tomatsu, S. CRISPR/nCas9-Edited CD34+ Cells Rescue Mucopolysaccharidosis IVA Fibroblasts Phenotype. Int. J. Mol. Sci. 2025, 26, 4334. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, S.G.; Poletto, E.; Lucot, K.L.; Colella, P.; Sheikali, A.; Montine, T.J.; Porteus, M.H.; Gomez-Ospina, N. Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat. Commun. 2020, 11, 3327. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Kumar, A.; Krug, S.; Mohan, H.; Rao, D.N.; Bishai, W.R.; Chandrasegaran, S. CRISPR Correction of the GBA Mutation in Human-Induced Pluripotent Stem Cells Restores Normal Function to Gaucher Macrophages and Increases Their Susceptibility to Mycobacterium tuberculosis. J. Infect. Dis. 2023, 228, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Scharenberg, S.G.; Camarena, J.; Kildebeck, E.J.; Clark, J.T.; Martin, R.M.; Bak, R.O.; Tang, Y.; Dohse, M.; Birgmeier, J.A.; et al. CRISPR/Cas9 Genome Engineering in Engraftable Human Brain-Derived Neural Stem Cells. iScience 2019, 15, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.B.; Seo, D.; Do, H.-S.; Han, Y.-M. Generation of a CRISPR/Cas9-corrected-hiPSC line (DDLABi001-A) from Fabry disease (FD)-derived iPSCs having α-galactosidase (GLA) gene mutation (c.803_806del). Stem Cell Res. 2023, 66, 103001. [Google Scholar] [CrossRef] [PubMed]
- Karl-Schöller, F.; Breyer, M.; Klopocki, E.; Üçeyler, N. Generation of a gene-corrected human isogenic iPSC line from a patient with Fabry disease carrying the GLA variant c.1069C>T using CRISPR/Cas9-mediated homology directed repair. Stem Cell Res. 2025, 86, 103711. [Google Scholar] [CrossRef] [PubMed]
- Rintz, E.; Banacki, M.; Ziemian, M.; Kobus, B.; Wegrzyn, G. Causes of death in mucopolysaccharidoses. Mol. Genet. Metab. 2024, 142, 108507. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Benincore-Flórez, E.; Rintz, E.; Herreño-Pachón, A.M.; Celik, B.; Ago, Y.; Alméciga-Díaz, C.J.; Tomatsu, S. Muco-polysaccharidoses: Cellular Consequences of Glycosaminoglycans Accumulation and Potential Targets. Int. J. Mol. Sci. 2022, 24, 477. [Google Scholar] [CrossRef] [PubMed]
- Consiglieri, G.; Bernardo, M.E.; Brunetti-Pierri, N.; Aiuti, A. Ex Vivo and In Vivo Gene Therapy for Mucopolysaccharidoses: State of the Art. Hematol. Oncol. Clin. N. Am. 2022, 36, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Alméciga-Díaz, C.J.; Tomatsu, S. Mucopolysaccharidosis IVA: Current Disease Models and Drawbacks. Int. J. Mol. Sci. 2023, 24, 16148. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; González, J.V.Á.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, G.N.; Gündüz, E. Gaucher Disease for Hematologists. Turk. J. Haematol. 2022, 39, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Daykin, E.C.; Ryan, E.; Sidransky, E. Diagnosing neuronopathic Gaucher disease: New considerations and challenges in assigning Gaucher phenotypes. Mol. Genet. Metab. 2021, 132, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; Maniwang, E.; Lopez, G.; Moaven, N.; Goldin, E.; Marugan, J.; Patnaik, S.; Dutra, A.; Southall, N.; et al. Macrophage Models of Gaucher Disease for Evaluating Disease Pathogenesis and Candidate Drugs. Sci. Transl. Med. 2014, 6, 240ra73. [Google Scholar] [CrossRef] [PubMed]
- Şoroğlu, C.V.; Berkay, E.G. Old disease-New reflections: Gaucher, immunity, and inflammation. J. Cell Mol. Med. 2024, 28, e70087. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hale, V.L.; Lelieveld, L.T.; Whitworth, L.J.; Busch-Nentwich, E.M.; Troll, M.; Edelstein, P.H.; Cox, T.M.; Roca, F.J.; Aerts, J.M.F.G.; et al. Gaucher disease protects against tuberculosis. Proc. Natl. Acad. Sci. USA 2023, 120, 2217673120. [Google Scholar] [CrossRef] [PubMed]
- Maghazachi, A. Globoid Cell Leukodystrophy (Krabbe Disease): An Update. ImmunoTargets Ther. 2023, 12, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Soloway, S.; Lister, D. Fabry’s Disease. N. Engl. J. Med. 2024, 391, 1038. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Chang, L.; Chen, H.; Gao, X.; Yin, J.; Zuo, Y.; Huang, Y.; Zhang, B.; Hu, J.; Liu, T. Improving the efficiency of CRISPR-Cas12a-based genome editing with site-specific covalent Cas12a-crRNA conjugates. Mol. Cell 2021, 81, 4747–4756.e7. [Google Scholar] [CrossRef] [PubMed]
- Mohr, M.; Damas, N.; Gudmand-Høyer, J.; Zeeberg, K.; Jedrzejczyk, D.; Vlassis, A.; Morera-Gómez, M.; Pereira-Schoning, S.; Puš, U.; Oliver-Almirall, A.; et al. The CRISPR-Cas12a Platform for Accurate Genome Editing, Gene Disruption, and Efficient Transgene Integration in Human Immune Cells. ACS Synth. Biol. 2023, 12, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Newby, G.A.; Yen, J.S.; Woodard, K.J.; Mayuranathan, T.; Lazzarotto, C.R.; Li, Y.; Sheppard-Tillman, H.; Porter, S.N.; Yao, Y.; Mayberry, K.; et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021, 595, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Everette, K.A.; Newby, G.A.; Levine, R.M.; Mayberry, K.; Jang, Y.; Mayuranathan, T.; Nimmagadda, N.; Dempsey, E.; Li, Y.; Bhoopalan, S.V.; et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat. Biomed. Eng. 2023, 7, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Byambaa, S.; Uosaki, H.; Ohmori, T.; Hara, H.; Endo, H.; Nureki, O.; Hanazono, Y. Non-viral ex vivo genome-editing in mouse bona fide hematopoietic stem cells with CRISPR/Cas9. Mol. Ther. Methods Clin. Dev. 2021, 20, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, H.; Smith, R.H.; Hakami, W.; Larochelle, A. Genome editing in human hematopoietic stem and progenitor cells via CRISPR-Cas9-mediated homology-independent targeted integration. Mol. Ther. 2021, 29, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Han, X.; Bai, X.; Yu, J.; Ma, Y.; Chen, W.; Zhang, D.; Li, Z. Revolutionizing cancer treatment: Enhancing CAR-T cell therapy with CRISPR/Cas9 gene editing technology. Front. Immunol. 2024, 15, 1354825. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Ceja, M.A.; Khericha, M.; Harris, C.M.; Puig-Saus, C.; Chen, Y.Y. CAR-T cell manufacturing: Major process parameters and next-generation strategies. J. Exp. Med. 2024, 221, 20230903. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maus, M.V.; Hinrichs, C.S. CAR T Cells and T-Cell Therapies for Cancer: A Translational Science Review. JAMA 2024, 332, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Chohan, K.L.; Siegler, E.L.; Kenderian, S.S. CAR-T Cell Therapy: The Efficacy and Toxicity Balance. Curr. Hematol. Malign-Rep. 2023, 18, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Rollo, B.; Sumer, H.; Cromer, B. Targeting the AAVS1 Site by CRISPR/Cas9 with an Inducible Transgene Cassette for the Neuronal Differentiation of Human Pluripotent Stem Cells. Methods Mol. Biol. 2022, 2495, 99–114. [Google Scholar] [PubMed]
- Leal, A.F.; Alméciga-Díaz, C.J. Efficient CRISPR/Cas9 nickase-mediated genome editing in an in vitro model of mucopoly-saccharidosis IVA. Gene Ther. 2022, 30, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Cifuentes, J.; Torres, C.E.; Suárez, D.; Quezada, V.; Gómez, S.C.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Alméciga-Díaz, C.J. Delivery and assessment of a CRISPR/nCas9-based genome editing system on in vitro models of mucopolysaccharidoses IVA assisted by magnetite-based nanoparticles. Sci. Rep. 2022, 12, 15045. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Cifuentes, J.; Quezada, V.; Benincore-Flórez, E.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Alméciga-Díaz, C.J. CRISPR/nCas9-Based Genome Editing on GM2 Gangliosidoses Fibroblasts via Non-Viral Vectors. Int. J. Mol. Sci. 2022, 23, 10672. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, S.; Chen, Y.-C.; Takemoto, T.; Sawatsubashi, S. Cas12a and MAD7, genome editing tools for breeding. Breed. Sci. 2024, 74, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Aznar, M.A.; Rech, A.J.; Good, C.R.; Kuramitsu, S.; Da, T.; Gohil, M.; Chen, L.; Hong, S.-J.A.; Ravikumar, P.; et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity 2023, 56, 2388–2407.e9. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.M.; Irfan, H.M.; Fatima, E.M.; Nazir, Z.M.; Verma, A.M.; Akilimali, A. Revolutionary breakthrough: FDA approves CASGEVY, the first CRISPR/Cas9 gene therapy for sickle cell disease. Ann. Med. Surg. 2024, 86, 4555–4559. [Google Scholar] [CrossRef] [PubMed]
- Kerwash, E.; Sajic, M.; Rantell, K.R.; McBlane, J.W.; Johnston, J.D.; Niewiarowska, A.; Butler, A.S.; Cole, S. Regulatory Assessment of Casgevy for the Treatment of Transfusion-Dependent β-Thalassemia and Sickle Cell Disease with Recurrent Vaso-Occlusive Crises. Curr. Issues Mol. Biol. 2024, 46, 8209–8225. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Babu, S.; Phan, M.; Yuan, S. CRISPR/Cas9-Based Protocol for Precise Genome Editing in Induced Pluripotent Stem Cells. Bio-Protocol. 2024, 14, e5141. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Chen, S.; Hsiao, S.; Jiang, Y.; Yang, Y.; Zhang, Y.; Wang, X.; Lai, Y.; Bauer, D.E.; Wu, Y. Therapeutic adenine base editing of human hematopoietic stem cells. Nat. Commun. 2023, 14, 207. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.-Y.; Thrasher, A.J. Gene therapy for X-linked severe combined immunodeficiency: Historical outcomes and current status. J. Allergy Clin. Immunol. 2020, 146, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegué, E. Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| * Cas Variant | MW (kDa) | Substrate Preference | PAM Requirement | Collateral Activity | Editing Mechanism |
|---|---|---|---|---|---|
| Cas9 | 160 | dsDNA | 5′-NGG-3′ | No | Blunt-end DSBs |
| Cas3 | 120 | ssDNA | Varies (Cascade-dependent) | No | Processive DNA degradation |
| Cas12a (Cpf1) | 130 | dsDNA | 5′-TTTV-3′ | Yes (ssDNA) | Staggered DSBs |
| Cas 12e (CasX) | 112 | dsDNA | 5′-TTCN-3′ | No | ssDNA cleavage |
| Cas12j (CasΦ) | 95 | ssDNA | PAM-independent | Yes (ssDNA) | Staggered DSBs |
| Cas12f1 | 60 | ssDNA | T-rich | Yes (ssDNA) | ssDNA cleavage |
| Cas14 | 50 | ssDNA | PAM-independent | Yes (ssDNA) | ssDNA cleavage |
| Cas7-11 | 120 | ssRNA | PFS-like motifs | No | RNA cleavage |
| Cas13 | 150 | ssRNA | PFS | Yes (ssRNA) | RNA cleavage |
| Model | CRISPR/Cas System Tested | Cell Type | Locus | IE (%) | OT (%) | Eng. (%) | Stemness | Ref. |
|---|---|---|---|---|---|---|---|---|
| MPS I | CRISPR/Cas9 | iPSCs | IDUA | 2.7 | NR | NA | Preserved | [71] |
| MPS I | CRISPR/Cas9 | HSPCs | CCR5 | 54 | <0.5 | 21.5 | Preserved | [72] |
| MPS I | CRISPR/Cas9 | HSPCs | CCR5 | TBI:5.7 BU:20 | NR | * TBI:45 * BU:73 | NR | [20] |
| MPS IVA | CRISPR/nCas9 | HSPCs | AAVS1 | 10 | ND | NA | Preserved | [73] |
| Gaucher | CRISPR/Cas9 | HSPC | CCR5 | 29.9 | NR | 23.2 | Preserved | [74] |
| Gaucher | CRISPR/Cas9 | iPSCs | GBA | NR | ND | NA | Preserved | [75] |
| Krabbe | CRISPR/Cas9 | NSC | IL2RG | 2.8 | <0.7 | NA | Preserved | [76] |
| Krabbe | CRISPR/Cas9 | NSC | CCR5 | 2.4 | NR | NA | NR | |
| Fabry | CRISPR/Cas9 | iPSCs | GLA | NR | NR | NA | Preserved | [77] |
| Fabry | CRISPR/Cas9 | iPSCs | GLA | NR | ND | NA | Preserved | [78] |
| Model | CRISPR/Cas System Tested | Cell Type | Locus | IE (%) | OT (%) | Eng. (%) | Stemness | Ref. |
|---|---|---|---|---|---|---|---|---|
| CAR-T | CRISPR/wtCas12 | T cells | TCR | 30 | NR | NA | NA | [91] |
| CRISPR/cCas12 | 52.7 | |||||||
| CAR-T | CRISPR/MAD7 | T cells | AAVS1 | 85 | ND | NA | NA | [92] |
| * AAVS1 | 30 | |||||||
| SCD | ABE | HSPCs | HBB | 80 | <2 | 70 | Preserved | [93] |
| SCD | PE3max | HSPCs | HBB | 41 | <1 | 97 | Preserved | [94] |
| X-SCID | HITI | HSPCs | IL2RG | 18.7 | ND | 13.2 | Preserved | [95] |
| LAD-1 | HITI | HSPCs | ITGB2 | 12 | ND | 21 | Preserved | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, A.F.; Prieto, L.E.; Pachajoa, H. CRISPR/Cas-Based Ex Vivo Gene Therapy and Lysosomal Storage Disorders: A Perspective Beyond Cas9. Cells 2025, 14, 1147. https://doi.org/10.3390/cells14151147
Leal AF, Prieto LE, Pachajoa H. CRISPR/Cas-Based Ex Vivo Gene Therapy and Lysosomal Storage Disorders: A Perspective Beyond Cas9. Cells. 2025; 14(15):1147. https://doi.org/10.3390/cells14151147
Chicago/Turabian StyleLeal, Andrés Felipe, Luis Eduardo Prieto, and Harry Pachajoa. 2025. "CRISPR/Cas-Based Ex Vivo Gene Therapy and Lysosomal Storage Disorders: A Perspective Beyond Cas9" Cells 14, no. 15: 1147. https://doi.org/10.3390/cells14151147
APA StyleLeal, A. F., Prieto, L. E., & Pachajoa, H. (2025). CRISPR/Cas-Based Ex Vivo Gene Therapy and Lysosomal Storage Disorders: A Perspective Beyond Cas9. Cells, 14(15), 1147. https://doi.org/10.3390/cells14151147