

Molecular Mechanisms and Pathways in Visceral Pain
Abstract
1. Chronic Visceral Pain: Mechanisms and Overlapping Pathologies
2. Pathophysiology of Visceral Pain and Hypersensitivity
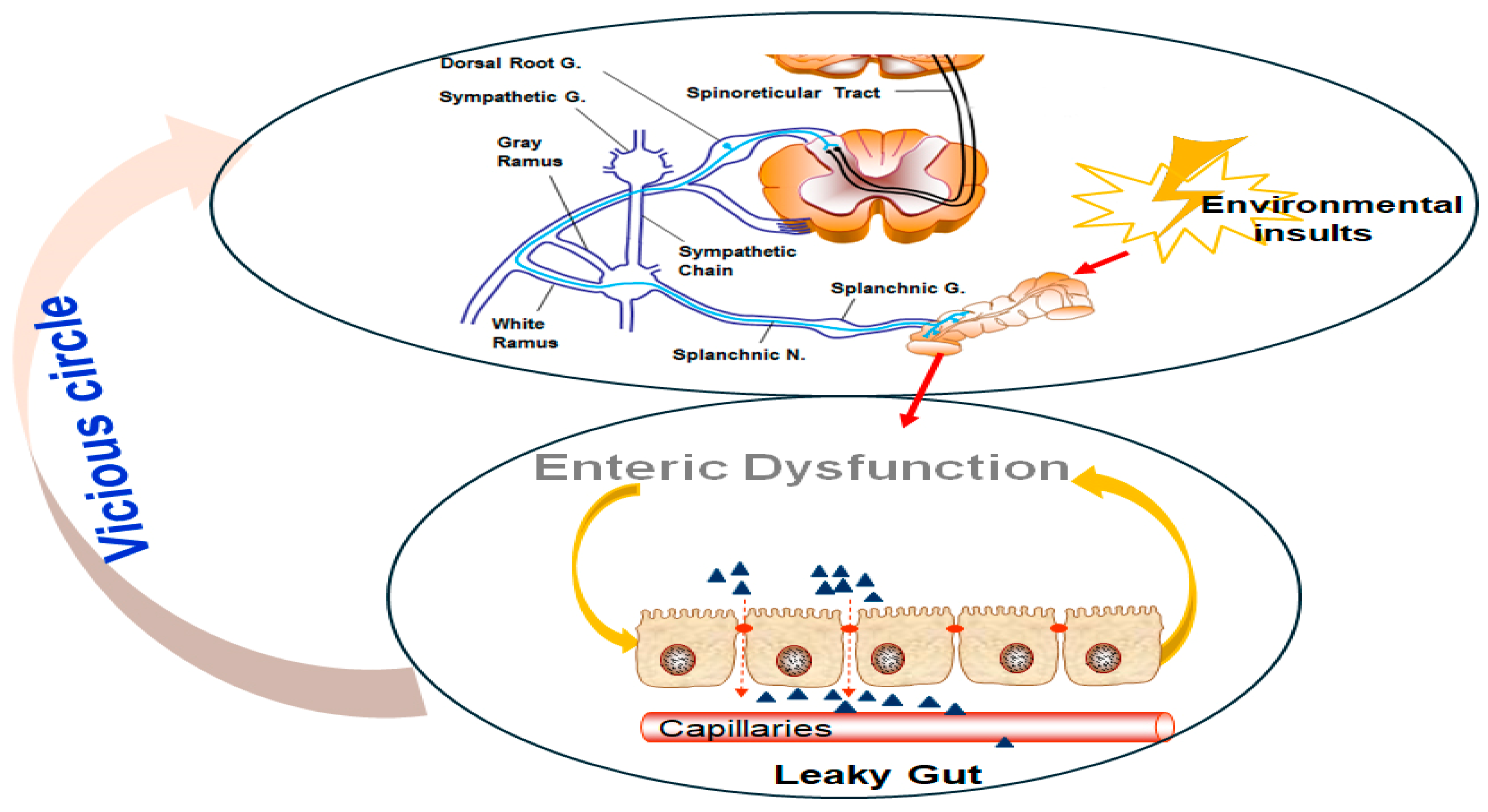
- Neuronal sensitization: Persistent, abnormal stimuli from the colon can lead to prolonged hypersensitivity by sensitizing spinal neurons [8,9]. This process involves viscerosomatic convergence, where nociceptive input from visceral structures (e.g., the gut and other internal organs) overlaps with somatic input (e.g., from the skin, muscles, and soft tissues), leading to a compound response. The sustained input from the colon can “prime” spinal neurons, making them more responsive to future stimuli [9].
- Increased intestinal permeability: The gut barrier is crucial for maintaining immune homeostasis and protecting against harmful stimuli [10]. However, barrier disruptions, such as those seen in conditions like IBS, can contribute to pain by allowing pathogenic or inflammatory mediators (e.g., cytokines, bacteria) to interact with afferent nerve fibers [6]. This “leaky gut” phenomenon may not only heighten nociception but also activate immune responses that further sensitize visceral pathways [10].
- Epigenetic influences: Recent studies have illuminated the role of epigenetic regulation in the pathophysiology of visceral pain [11]. Altered expression of microRNAs (miRNAs) in GI tissues, potentially delivered via extracellular vesicles (EVs), may affect the expression of pain-related genes. These small RNA molecules can modulate pain signaling pathways at both the peripheral and central levels, adding another layer of complexity to visceral hypersensitivity mechanisms.
2.1. Mechanisms of Neuronal Sensitization
2.1.1. Afferent Mechanisms of Visceral Pain
2.1.2. Central Sensitization and Viscerosomatic Convergence
2.1.3. Overlap with Chronic Pelvic Pain Disorders
2.2. Intestinal Barrier Dysfunction and Visceral Pain
2.3. Epigenetic Mechanisms in Visceral Pain
3. Key Molecular Mediators and Possible Targets for Treatment of Visceral Pain
3.1. Neurotransmitters and Neuromodulators in Pain Signaling
3.1.1. Glutamate and Gamma-Aminobutyric Acid (GABA)
3.1.2. Substance P and Calcitonin Gene-Related Peptide (CGRP)
3.1.3. Serotonin (5-HT): Receptor Subtypes
3.1.4. Transient Receptor Potential Channels (TRP)
3.1.5. Voltage-Gated Sodium Channels
3.1.6. Catechol-O-Methyltransferase (COMT)
3.1.7. Ion Channels
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASIC | Acid-sensing ion channel |
| CGRP | Calcitonin gene-related peptide |
| circRNA | Circular ribonucleic acid |
| CNS | Central nervous system |
| COMT | Catechol-O-methyltransferase |
| CUGBP1 | CUG triplet repeat RNA-binding protein 1 |
| DGBI | Disorder of gut–brain interaction |
| DRA | Downregulated in adenoma protein |
| DRG | Dorsal root ganglia |
| EV | Extracellular vesicle |
| FGF | Fibroblast growth factor |
| FGID | Functional gastrointestinal disorder |
| GABA | Gamma-aminobutyric acid |
| GI | Gastrointestinal |
| HIF-1α | Hypoxia-inducible factor 1 subunit alpha |
| IBS | Irritable bowel syndrome |
| IBS-C | IBS with constipation |
| IL | Interleukin |
| lncRNA | Long non-coding RNA |
| IJ | Intercellular junction |
| KO | Knockout |
| LTP | Long-term potentiation |
| miRNA | MicroRNA |
| mRNA | Messenger RNA |
| ncRNA | Non-coding RNA |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHE3 | Sodium/hydrogen exchanger isoform 3 |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| PAMPs | Pathogen-associated molecular patterns |
| PI3K | Phosphatidylinositol 3 kinase |
| PI-IBS | Post-infectious IBS |
| PI-IBS-C | PI-IBS with constipation |
| PI-IBS-D | PI-IBS with diarrhea |
| ROS | Reactive oxygen species |
| SHIP-1 | Src homology 2 domain-containing inositol phosphatase 1 |
| TGF-β | Transforming growth factor beta |
| TJ | Tight junction |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-alpha |
| TRPA1 | Transient receptor potential ankyrin 1 |
| TRPV1 | Transient receptor potential vanilloid 1 |
| vtRNA | Vault RNA |
References
- Videlock, E.J.; Chang, L. Latest Insights on the Pathogenesis of Irritable Bowel Syndrome. Gastroenterol. Clin. N. Am. 2021, 50, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Ryu, H.J.; Bhatt, R.R. The Neurobiology of Irritable Bowel Syndrome. Mol. Psychiatry 2023, 28, 1451–1465. [Google Scholar] [CrossRef] [PubMed]
- Keefer, L.; Ballou, S.K.; Drossman, D.A.; Ringstrom, G.; Elsenbruch, S.; Ljótsson, B. A Rome Working Team Report on Brain-Gut Behavior Therapies for Disorders of Gut-Brain Interaction. Gastroenterology 2022, 162, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Price, D.D.; Callam, C.S.; Woodruff, M.A.; Verne, G.N. Effects of the N-Methyl-D-Aspartate Receptor on Temporal Summation on Second Pain (Wind-up) in Irritable Bowel Syndrome. J. Pain 2011, 12, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Hong, S.; Wiley, J.W. The Role of Epigenomic Regulatory Pathways in the Gut-Brain Axis and Visceral Hyperalgesia. Cell Mol. Neurobiol. 2022, 42, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Grover, M.; Vanuytsel, T.; Chang, L. Intestinal Permeability in Disorders of Gut-Brain Interaction (DGBI): From Bench to Bedside. Gastroenterology 2024, 168, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Drossman, D.A.; Tack, J. Rome Foundation Clinical Diagnostic Criteria for Disorders of Gut-Brain Interaction. Gastroenterology 2022, 162, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Hockley, J.R.F.; Taylor, T.S.; Callejo, G.; Wilbrey, A.L.; Gutteridge, A.; Bach, K.; Winchester, W.J.; Bulmer, D.C.; McMurray, G.; Smith, E.S.J. Single-cell RNA-seq reveals seven classes of colonic sensory neuron. Gut 2019, 68, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Brierley, S.M.; Hibberd, T.J.; Spencer, N.J. Spinal afferent innervation of the colon and rectum. Front. Cell Neurosci. 2018, 12, 467. [Google Scholar] [CrossRef] [PubMed]
- Edogawa, S.; Edwinson, A.L.; Peters, S.A.; Chikkamenahalli, L.L.; Sundt, W.; Graves, S.; Gurunathan, S.V.; Breen-Lyles, M.; Johnson, S.; Dyer, R.; et al. Serine proteases as luminal mediators of intestinal barrier dysfunction and symptom severity in IBS. Gut 2020, 69, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Mahurkar-Joshi, S.; Chang, L. Epigenetic Mechanisms in Irritable Bowel Syndrome. Front. Psychiatry 2020, 11, 805. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Q.; Price, D.D.; Verne, G.N. Reversal of Visceral and Somatic Hypersensitivity in a Subset of Hypersensitive Rats by Intracolonic Lidocaine. Pain 2008, 139, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Latremoliere, A.; Woolf, C.J. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed]
- Inquimbert, P.; Moll, M.; Latremoliere, A.; Tong, C.-K.; Whang, J.; Sheehan, G.F.; Smith, B.M.; Korb, E.; Athié, M.C.; Babaniyi, O.; et al. NMDA Receptor Activation Underlies the Loss of Spinal Dorsal Horn Neurons and the Transition to Persistent Pain after Peripheral Nerve Injury. Cell Rep. 2018, 23, 2678–2689. [Google Scholar] [CrossRef] [PubMed]
- Verne, G.N.; Sen, A.; Price, D.D. Intrarectal lidocaine is an effective treatment for abdominal pain associated w diarrhea-predominant irritable bowel syndrome. J. Pain 2005, 6, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Young, R.L.; Kaelberer, M.M.; Cevikbas, V.; Berthoud, H.R.; Schneider, S.; Brierley, S.M. The Role of Enteroendocrine-Sensory Cell Communication in Visceral Nociception. J. Neurochem. 2023, 164, 748. [Google Scholar]
- Ren, K.; Dubner, R. Molecular Mechanisms Underlying Nociception and Pain Plasticity: Bridging Peripheral and Central Mechanisms. Nat. Rev. Neurosci. 2024, 25, 189–205. [Google Scholar]
- Woolf, C.J. Central Sensitization: Implications for the Diagnosis and Treatment of Pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef] [PubMed]
- Gewandter, J.S.; Chaudari, J.; Iwan, K.B.; Kitt, R.; As-Sanie, S.; Bachmann, G.; Clemens, Q.; Lai, H.H.; Tu, F.; Verne, G.N.; et al. Research Design Characteristics of Published Pharmacologic Randomized Clinical Trials for Irritable Bowel Syndrome and Chronic Pelvic Pain Conditions: An ACTTION Systematic Review. J. Pain 2018, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Atmani, K.; Wuestenberghs, F.; Baron, M.; Bouleté, I.; Guérin, C.; Bahlouli, W.; Vaudry, D.; de Rego, J.C.; Cornu, J.N.; Leroi, A.M.; et al. Bladder–colon chronic cross-sensitization involves neuro-glial pathways in male mice. World J. Gastroenterol. 2022, 28, 6935–6949. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Q.; Verne, M.L.; Fields, J.Z.; Lefante, J.J.; Basra, S.; Salameh, H.; Verne, G.N. Randomised Placebo-Controlled Trial of Dietary Glutamine Supplements for Postinfectious Irritable Bowel Syndrome. Gut 2019, 68, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Zhou, Y.; Turner, J.R. Tight Junction Regulation, Intestinal Permeability, and Mucosal Immunity in Gastrointestinal Health and Disease. Curr. Opin. Gastroenterol. 2024, 41, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Priyamvada, S.; Ge, Y.; Jayawardena, D.; Singhal, M.; Anbazhagan, A.N.; Chatterjee, I.; Dayal, A.; Patel, M.; Zadeh, K.; et al. A Novel Role of SLC26A3 in Maintenance of Intestinal Epithelial Barrier Integrity. Gastroenterology 2020, 160, 1240–1255.e3. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla, N.; Qazi, A.; Sharma, A.; Mongan, K.; Comiskey, S.; Manne, J.; Youkhana, A.G.; Khanna, S.; Saksena, S.; Dudeja, P.K.; et al. Mucosal Metabolomic Signatures in Chronic Colitis: Novel Insights into the Pathophysiology of Inflammatory Bowel Disease. Metabolites 2023, 13, 873. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xiao, L.; Chung, H.K.; Chen, T.; Mallard, C.G.; Warner, B.; Yu, T.X.; Kwon, M.S.; Chae, S.; Raufman, J.P.; et al. Noncoding Vault RNA1-1 Impairs Intestinal Epithelial Renewal and Barrier Function by Interacting With CUG-Binding Protein 1. Cell. Mol. Gastroenterol. Hepatol. 2025, 19, 101410. [Google Scholar] [CrossRef] [PubMed]
- Mahurkar-Joshi, S.; Rankin, C.R.; Videlock, E.J.; Soroosh, A.; Verma, A.; Khandadash, A.; Iliopoulos, D.; Pothoulakis, C.; Mayer, E.A.; Chang, L. The Colonic Mucosal MicroRNAs, MicroRNA-219a-5p, and MicroRNA-338-3p Are Downregulated in Irritable Bowel Syndrome and Are Associated With Barrier Function and MAPK Signaling. Gastroenterology 2021, 160, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Q.; Souba, W.W.; Croce, C.M.; Verne, G.N. MicroRNA-29a Regulates Intestinal Membrane Permeability in Patients with Irritable Bowel Syndrome. Gut 2010, 59, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Q.; Costinean, S.; Croce, C.M.; Brasier, A.R.; Merwat, S.; Larson, S.A.; Basra, S.; Verne, G.N. MicroRNA 29 Targets Nuclear Factor-ΚB-Repressing Factor and Claudin 1 to Increase Intestinal Permeability. Gastroenterology 2015, 148, 158–169.e8. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.K.; Xiao, L.; Han, N.; Chen, J.; Yao, V.; Cairns, C.M.; Raufman, B.; Rao, J.N.; Turner, D.J.; Kozar, R.; et al. Circular RNA Cdr1as Inhibits Proliferation and Delays Injury-Induced Regeneration of the Intestinal Epithelium. JCI Insight 2024, 9, e169716. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-Regulation of MIR200C-3p, Which Degrades Occludin MRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Freire, V.S.; Burkhard, F.C.; Kessler, T.M.; Kuhn, A.; Draeger, A.; Monastyrskaya, K. MicroRNAs May Mediate the Down-Regulation of Neurokinin-1 Receptor in Chronic Bladder Pain Syndrome. Am. J. Pathol. 2010, 176, 288–303. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yang, C.; Kirkmire, C.M.; Wang, Z.J. Regulation of Opioid Tolerance by Let-7 Family MicroRNA Targeting the μ Opioid Receptor. J. Neurosci. 2010, 30, 10251–10258. [Google Scholar] [CrossRef] [PubMed]
- Schank, J.R.; Heilig, M. Substance P and the Neurokinin-1 Receptor: The New Corticotropin-Releasing Factor. Int. Rev. Neurobiol. 2017, 136, 151–175. [Google Scholar] [PubMed]
- Brierley, S.; Linden, D. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Alves, N.D.; Del Colle, A.; Talati, A.; Najjar, S.A.; Bouchard, V.; Gillet, V.; Tong, Y.; Huang, Z.; Browning, K.N.; et al. Intestinal epithelial serotonin as a novel target for treating disorders of gut–brain interaction and mood. Gastroenterology 2025, 168, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.N.; Nakamura, M.; Kawashima, H. New role of serotonin as a biomarker of gut–brain interaction. Life 2023, 14, 1280. [Google Scholar] [CrossRef] [PubMed]
- Julius, D. TRP Channels and Pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, L.; Larson, S.; Basra, S.; Merwat, S.; Tan, A.; Croce, C.; Nicholas Verne, G. Decreased MiR-199 Augments Visceral Pain in Patients with IBS through Translational Upregulation of TRPV1. Gut 2015, 65, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Sampson, K.; Iyer, V.; Julius, D.; Bosmans, F. Pharmacology of the Nav1.1 Domain IV Voltage Sensor Reveals Coupling between Inactivation Gating Processes. Proc. Natl. Acad. Sci. USA 2017, 114, 6836–6841. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective Spider Toxins Reveal a Role for the Nav1.1 Channel in Mechanical Pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Meloto, C.B.; Segall, S.K.; Smith, S.; Parisien, M.; Shabalina, S.A.; Rizzatti-Barbosa, C.M.; Gauthier, J.; Tsao, D.; Convertino, M.; Piltonen, M.H.; et al. COMT Gene Locus: New Functional Variants. Pain 2015, 156, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Vase, L.; Petersen, G.L.; Riley JL3rd Price, D.D. COMT Val158Met genotype modulates μ-opioid system activation during sustained pain and placebo analgesia. Pain Rep. 2019, 4, e780. [Google Scholar]
- Zhou, Q.Q.; Yang, L.; Verne, M.L.; Zhang, B.B.; Fields, J.; Verne, G.N. Catechol-O-Methyltransferase Loss Drives Cell-Specific Nociceptive Signaling via the Enteric Catechol-O-Methyltransferase/MicroRNA-155/Tumor Necrosis Factor α Axis. Gastroenterology 2023, 164, 630–641.e34. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Zhang, F.C.; Xu, T.W.; Weng, R.X.; Zhang, H.H.; Chen, Q.Q.; Hu, S.; Gao, R.; Li, R.; Xu, G.Y. Advances in the pathological mechanisms and clinical treatments of visceral pain. J. Neuroinflamm. 2024, 21, 115. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sayuk, G.S.; Rosenbaum, D.P.; Edelstein, S.; Kozuka, K.; Chang, L. An Overview of the Effects of Tenapanor on Visceral Hypersensitivity in the Treatment of Irritable Bowel Syndrome with Constipation. Clin. Exp. Gastroenterol. 2024, 17, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Harris, L.A.; Lacy, B.E.; Quigley, E.M.M.; Moayyedi, P. Systematic review with meta-analysis: The efficacy of prebiotics, probiotics, synbiotics and antibiotics in irritable bowel syndrome. Aliment. Pharmacol. Ther. 2023, 40, 187–214. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Neurotransmitter/ Neuromodulator | Mechanism | Role in Pain Signaling | Therapeutic Implications |
|---|---|---|---|
| Glutamate [22] | Primary excitatory neurotransmitter in the CNS; acts through NMDA, AMPA, and mGluRs | Mediates excitatory nociceptive signaling, contributing to central sensitization | Targeting glutamate receptors (NMDA, AMPA) and GABA receptors can help manage chronic pain and hypersensitivity, particularly in IBS |
| GABA [15] | Principal inhibitory neurotransmitter; acts through GABA-A and GABA-B receptors | Maintains neural homeostasis by inhibiting excessive excitatory signaling | |
| Substance P [36] | Released by primary sensory neurons; binding to NK1 receptors amplifies pain signals and mediates inflammation | Enhance pain transmission and contribute to neurogenic inflammation, facilitating peripheral and central sensitization | Potential targets for managing visceral pain syndromes, especially in IBS and related disorders |
| CGRP [35] | Promotes vasodilation and inflammation, often co-released with substance P | ||
| Serotonin (5-HT) [36,37] | 5-HT3 receptors: mediate excitatory transmission and contribute to visceral pain | 5-HT signaling regulates gut function and is implicated in conditions like IBS Dysregulated 5-HT3 signaling contributes to visceral hypersensitivity | Modulation of serotonin receptors, particularly 5-HT3 antagonists and 5- HT4 agonists, offers potential therapeutic strategies for gut disorders |
| 5-HT4 receptors: regulate GI motility and sensitivity | 5-HT signaling regulates gut function and is implicated in conditions like IBS 5-HT4 agonists improve motility and reduce pain | ||
| TRPV1 channels [38,39] | Activated by noxious stimuli, such as heat and acid | Contribute to inflammatory pain responses and hyperalgesia Lowers its activation threshold during inflammation | Antagonists show promise as novel therapies for visceral pain and hyperalgesia, particularly in inflammatory conditions |
| TRPA1 channels [38] | Activated by environmental irritants and inflammatory mediators | Contribute to inflammatory pain responses and hyperalgesia participates in inflammatory pain | |
| Voltage-gated sodium channels [40,41] | NaV1.7 and NaV1.8 channels facilitate action potential propagation in sensory neurons, with activity enhanced in pathological conditions like inflammation | Essential for pain signaling and contribute to neuronal hyperexcitability in chronic pain conditions | NaV channel blockers are potential pain management therapies, particularly for neuropathic and inflammatory pain |
| COMT [43,44] | COMT degrades catecholamines (dopamine, norepinephrine), modulating pain sensitivity | Genetic variants in COMT influence pain perception; higher COMT activity is associated with reduced pain sensitivity Inflammation can modulate COMT’s effects on pain signaling | Targeting COMT in combination with other therapies may reduce chronic pain, particularly in conditions like fibromyalgia and IBS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Verne, G.N. Molecular Mechanisms and Pathways in Visceral Pain. Cells 2025, 14, 1146. https://doi.org/10.3390/cells14151146
Zhou Q, Verne GN. Molecular Mechanisms and Pathways in Visceral Pain. Cells. 2025; 14(15):1146. https://doi.org/10.3390/cells14151146
Chicago/Turabian StyleZhou, Qiqi, and George Nicholas Verne. 2025. "Molecular Mechanisms and Pathways in Visceral Pain" Cells 14, no. 15: 1146. https://doi.org/10.3390/cells14151146
APA StyleZhou, Q., & Verne, G. N. (2025). Molecular Mechanisms and Pathways in Visceral Pain. Cells, 14(15), 1146. https://doi.org/10.3390/cells14151146