
The Role of Nuclear Phosphoinositides in the p53-MDM2 Nexus
 , , , and
, , , and {kind=link}
{kind=link}
Abstract
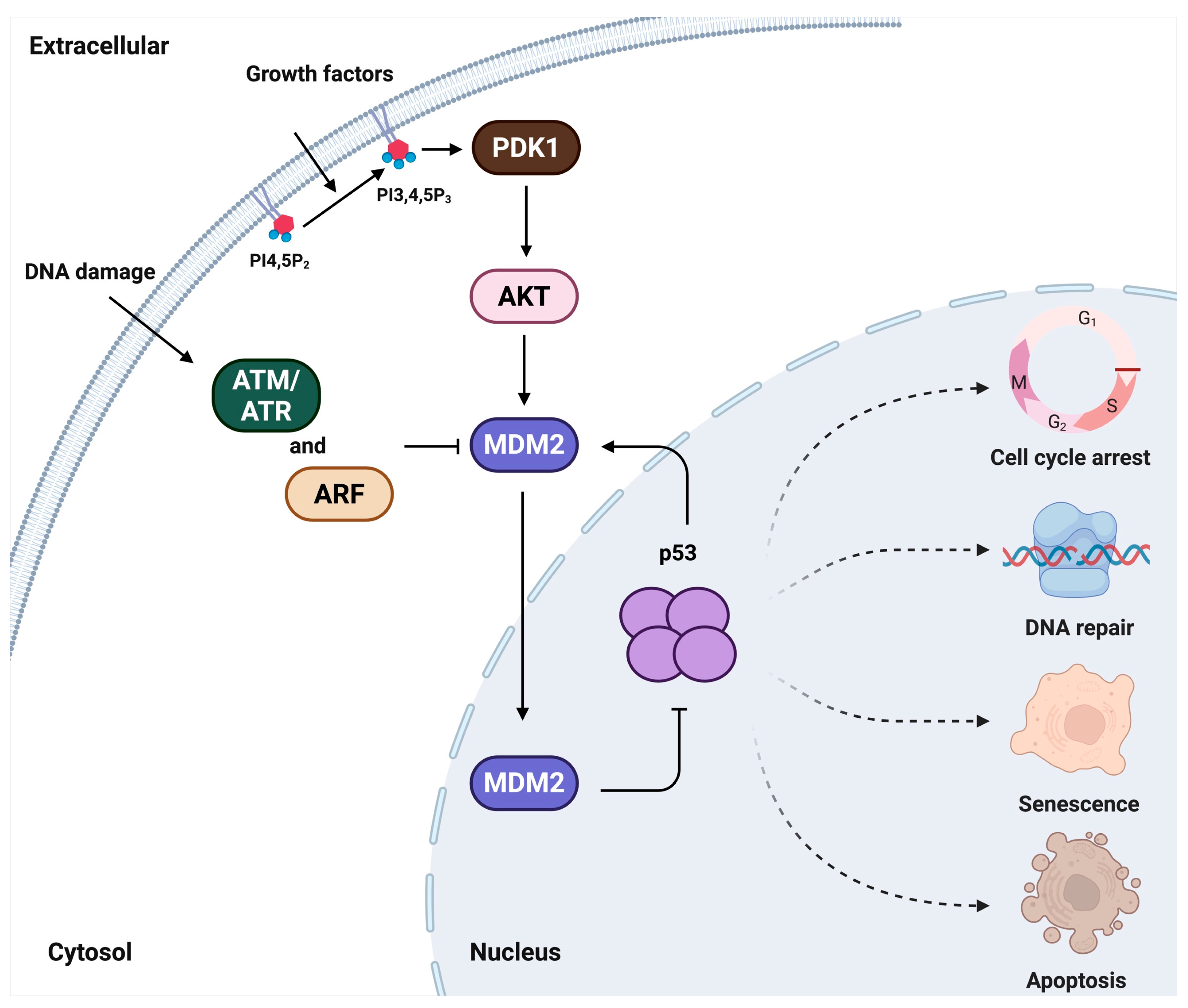
1. Introduction
2. p53
3. MDM2
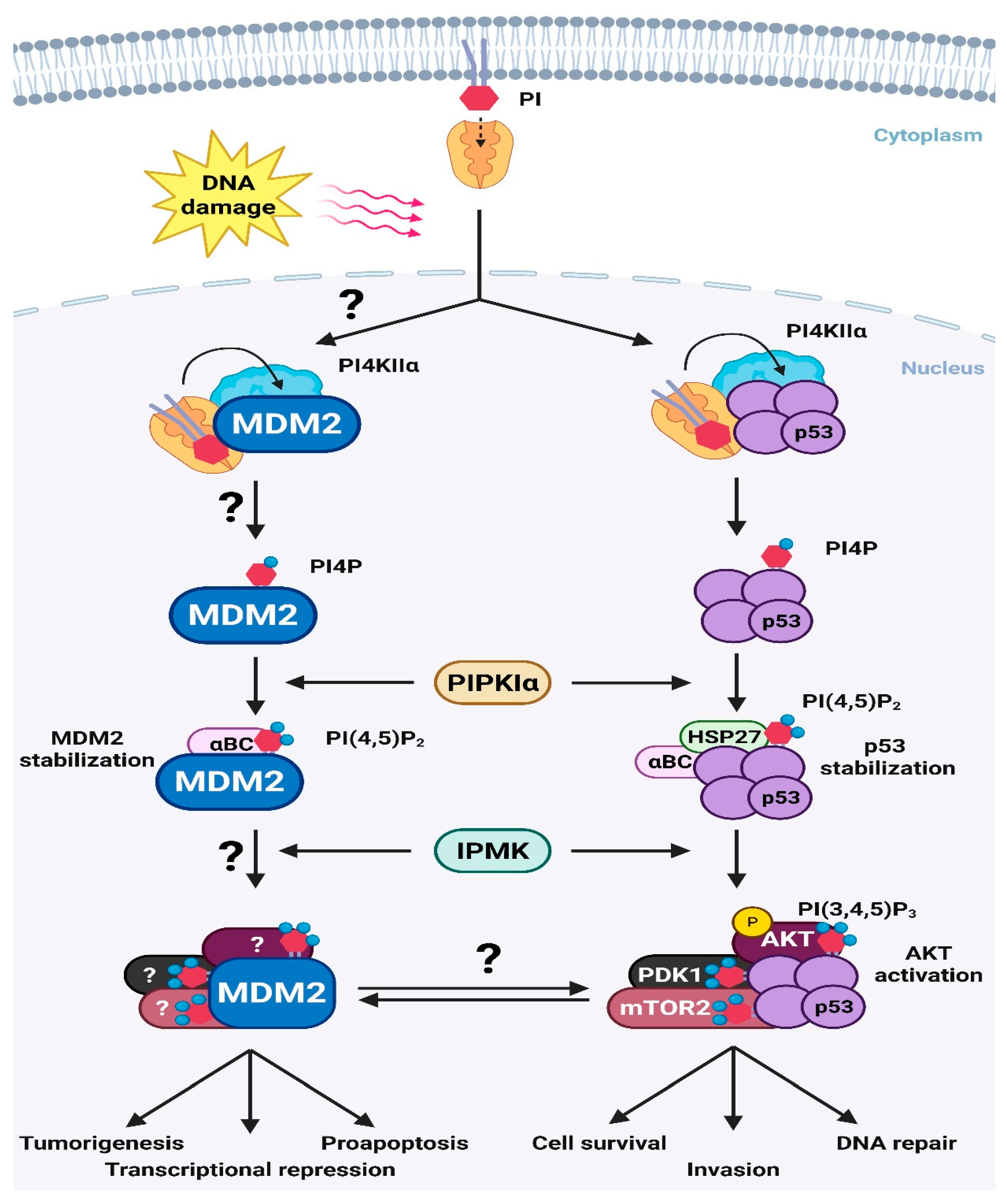
4. Potential Targets of the p53-MDM2-PIPn Nexus
4.1. Nuclear p53 and Its Targets
4.2. Nuclear AKT and Its Targets
5. p53-Independent Functions of MDM2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Boronenkov, I.V.; Loijens, J.C.; Umeda, M.; Anderson, R.A. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell 1998, 9, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.A.; Laishram, R.S.; Anderson, R.A. Nuclear phosphoinositides: A signaling enigma wrapped in a compartmental conundrum. Trends Cell Biol. 2012, 20, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Chen, M.; Cryns, V.L.; Anderson, R.A. A nuclear phosphoinositide kinase complex regulates p53. Nat. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Choi, S.; Wen, T.; Chen, C.; Thapa, N.; Lee, J.H.; Cryns, V.L.; Anderson, R.A. A p53-phosphoinositide signalosome regulates nuclear AKT activation. Nat. Cell Biol. 2022, 24, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Carrillo, N.D.; Chen, M.; Wen, T.; Awasthi, P.; Anderson, R.A.; Cryns, V.L. Regulation of NRF2 by stably associated phosphoinositides and small heat shock proteins in response to stress. J. Biol. Chem. 2025, 301, 110367. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Chen, M.; Cryns, V.L.; Anderson, R.A. The Poly(A) Polymerase Star-PAP is Regulated by Stably Associated Phosphoinositide Messengers. J. Biol. Chem. 2025, 301, 110412. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Chen, M.; Wen, T.; Anderson, R.A.; Cryns, V.L. Regulation of the MDM2-p53 Nexus by a Nuclear Phosphoinositide and Small Heat Shock Protein Complex. bioRxiv 2025. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, S.A.; O’Hagan, H.M.; Ljungman, M. Accumulation of soluble and nucleolar-associated p53 proteins following cellular stress. J. Cell Sci. 2001, 114 Pt 10, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Marine, J.C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ross, J.S.; Gay, L.; Dayyani, F.; Roszik, J.; Subbiah, V.; Kurzrock, R. Analysis of MDM2 Amplification: Next-Generation Sequencing of Patients With Diverse Malignancies. JCO Precis. Oncol. 2018, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, N.D.; Chen, M.; Wen, T.; Awasthi, P.; Wolfe, T.J.; Sterling, C.; Cryns, V.L.; Anderson, R.A. Lipid Transfer Proteins and PI4KIIα Initiate Nuclear p53-Phosphoinositide Signaling. bioRxiv 2025. [Google Scholar] [CrossRef]
- Irvine, R.F. Nuclear lipid signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Bunney, T.D.; Katan, M. Phosphoinositide signalling in cancer: Beyond PI3K and PTEN. Nat. Rev. Cancer 2010, 10, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Baek, M.-J.; Wooldrik, C.; Johnson, K.R.; Fisher, K.W.; Lou, J.; Ricks, T.J.; Wen, T.; Best, M.D.; Cryns, V.L.; et al. Nuclear phosphoinositide signaling promotes YAP/TAZ-TEAD transcriptional activity in breast cancer. EMBO J. 2024, 43, 1740–1769. [Google Scholar] [CrossRef] [PubMed]
- Blind, R.D.; Suzawa, M.; Ingraham, H.A. Direct modification and activation of a nuclear receptor-PIP2 complex by the inositol lipid kinase IPMK. Sci. Signal. 2012, 5, ra44. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Thapa, N.; Hedman, A.C.; Li, Z.; Sacks, D.B.; Anderson, R.A. IQGAP1 is a novel phosphatidylinositol 4,5 bisphosphate effector in regulation of directional cell migration. EMBO J. 2013, 32, 2617–2630. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Tong, D.R.; Manfredi, J.; Prives, C. The Tail That Wags the Dog: How the Disordered C-Terminal Domain Controls the Transcriptional Activities of the p53 Tumor-Suppressor Protein. Trends Biochem. Sci. 2016, 41, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wen, T.; Horn, H.T.; Chandrahas, V.K.; Thapa, N.; Choi, S.; Cryns, V.L.; Anderson, R.A. The nuclear phosphoinositide response to stress. Cell Cycle 2020, 19, 268–289. [Google Scholar] [CrossRef] [PubMed]
- Malin, D.; Petrovic, V.; Strekalova, E.; Sharma, B.; Cryns, V.L. αB-crystallin: Portrait of a malignant chaperone as a cancer therapeutic target. Pharmacol. Ther. 2016, 160, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.; Sarcevic, B.; Musgrove, E.A.; Watts, C.K.; Sutherland, R.L. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 1997, 272, 10882–10894. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Jacobs, S.B.R.; Krieg, A.J.; Pathak, N.; Zeng, Q.; Kaldis, P.; Giaccia, A.J.; Attardi, L.D. The Metastasis-Associated Gene Prl-3 Is a p53 Target Involved in Cell-Cycle Regulation. Mol. Cell 2008, 30, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rozenfeld, N.; et al. Mutant p53 Prolongs NF-κB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.J.; Wee, Z.N.; et al. PDK1 Signaling Toward PLK1–MYC Activation Confers Oncogenic Transformation, Tumor-Initiating Cell Activation, and Resistance to mTOR-Targeted Therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, D.; Shi, H.; Qian, H.; Chen, H.; Zeng, Q.; Jin, F.; Ye, Y.; Ou, Z.; Guo, M.; et al. PDK1 promotes breast cancer progression by enhancing the stability and transcriptional activity of HIF-1α. Genes Dis. 2024, 11, 101041. [Google Scholar] [CrossRef] [PubMed]
- Mahanivong, C.; Chen, H.M.; Yee, S.W.; Pan, Z.K.; Dong, Z.; Huang, S. Protein kinase C alpha-CARMA3 signaling axis links Ras to NF-kappa B for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells. Oncogene 2008, 27, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.L.; Bamlet, W.R.; Smyrk, T.C.; Fields, A.P.; Murray, N.R. Protein kinase Ciota is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Res. 2010, 70, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.M.; Scotti Buzhardt, M.L.; Li, S.; Smith, K.E.; Fields, A.P.; Murray, N.R. Protein kinase C zeta regulates human pancreatic cancer cell transformed growth and invasion through a STAT3-dependent mechanism. PLoS ONE 2013, 8, e72061. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, J.; Takagi, E.; Wang, F.; Saha, B.; Liu, X.; Joubert, L.-M.; Gleason, C.E.; Jin, M.; Li, C.; et al. Interactions between mTORC2 core subunits Rictor and mSin1 dictate selective and context-dependent phosphorylation of substrate kinases SGK1 and Akt. J. Biol. Chem. 2022, 298, 102288. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Matsui, T.; Novikov, M.; Park, J.; Hemmings, B.; Rosenzweig, A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation 2005, 111, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Wang, C.Y.; Guttridge, D.C.; Schottelius, A.J.; Baldwin, A.S., Jr.; Mayo, M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol. Cell. Biol. 2000, 20, 1626–1638. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Kutuk, O.; Brito, G.C.; Andrews, T.S.; Leber, B.; Letai, A.; Andrews, D.W. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 2018, 19, e45235. [Google Scholar] [CrossRef] [PubMed]
- Kizilboga, T.; Baskale, E.A.; Yildiz, J.; Akcay, I.M.; Zemheri, E.; Can, N.D.; Ozden, C.; Demir, S.; Ezberci, F.; Dinler-Doganay, G. Bag-1 stimulates Bad phosphorylation through activation of Akt and Raf kinases to mediate cell survival in breast cancer. BMC Cancer 2019, 19, 1254. [Google Scholar] [CrossRef] [PubMed]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.-Y.; Lai, C.-C.; Chang, C.-J.; Huang, W.-C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Cha, C.; Lewis, R.; Jhanwar, S.C.; Huvos, A.G.; Healey, J.H. MDM2 gene amplification in metastatic osteosarcoma. Cancer Res. 1993, 53, 16–18. [Google Scholar] [PubMed]
- Datta, M.W.; Macri, E.; Signoretti, S.; Renshaw, A.A.; Loda, M. Transition from in situ to invasive testicular germ cell neoplasia is associated with the loss of p21 and gain of mdm-2 expression. Mod. Pathol. 2001, 14, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Li, Z.; Broglio, K.; Berry, D.A.; Hung, M.C. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell. Biol. 2006, 26, 7269–7282. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Trouche, D.; Hagemeier, C.; Sørensen, T.S.; La Thangue, N.B.; Kouzarides, T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature 1995, 375, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Loughran, O.; La Thangue, N.B. Apoptotic and growth-promoting activity of E2F modulated by MDM2. Mol. Cell. Biol. 2000, 20, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; Lushnikova, T.; Plaza, S.; Eischen, C.M. Mdm2 promotes genetic instability and transformation independent of p53. Mol. Cell. Biol. 2008, 28, 4862–4874. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Girnita, A.; Larsson, O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 8247–8252. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Froment, P.; Dupont, J.; Christophe-Marine, J. Mdm2 exerts pro-apoptotic activities by antagonizing insulin-like growth factor-I-mediated survival. Cell Cycle 2008, 7, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Minsky, N.; Oren, M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol. Cell 2004, 16, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.; Nield, P.C.; Devling, T.; Jenkins, R.E.; Park, B.K.; Polański, R.; Vlatković, N.; Boyd, M.T. MDM2 regulates dihydrofolate reductase activity through monoubiquitination. Cancer Res. 2008, 68, 3232–3242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Hariharan, A.; Bastianello, G.; Toyama, Y.; Shivashankar, G.V.; Foiani, M.; Sheetz, M.P. DNA damage causes rapid accumulation of phosphoinositides for ATR signaling. Nat. Commun. 2017, 8, 2118. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Salah, M.K.; Chen, X.; Kucherenko, N.V.; Cryns, V.L.; Anderson, R.A. The Role of Nuclear Phosphoinositides in the p53-MDM2 Nexus. Cells 2025, 14, 1126. https://doi.org/10.3390/cells14151126
Lee JH, Salah MK, Chen X, Kucherenko NV, Cryns VL, Anderson RA. The Role of Nuclear Phosphoinositides in the p53-MDM2 Nexus. Cells. 2025; 14(15):1126. https://doi.org/10.3390/cells14151126
Chicago/Turabian StyleLee, Jeong Hyo, Muhammad Khalil Salah, Xiangqin Chen, Nickolas Vladimir Kucherenko, Vincent L. Cryns, and Richard A. Anderson. 2025. "The Role of Nuclear Phosphoinositides in the p53-MDM2 Nexus" Cells 14, no. 15: 1126. https://doi.org/10.3390/cells14151126
APA StyleLee, J. H., Salah, M. K., Chen, X., Kucherenko, N. V., Cryns, V. L., & Anderson, R. A. (2025). The Role of Nuclear Phosphoinositides in the p53-MDM2 Nexus. Cells, 14(15), 1126. https://doi.org/10.3390/cells14151126