
Carcinogenesis Associated with Toxin Nephropathy: Proposed Mediation by Phosphate Toxicity


Abstract
1. Introduction
“…any adverse functional or structural changes in the kidney due to the effect of a chemical or biological product that is inhaled, ingested, injected or otherwise absorbed, or that yields toxic metabolites with an identifiable adverse effect to the kidneys” [13].
2. Materials and Methods
3. Mechanisms of Nephrotoxicity
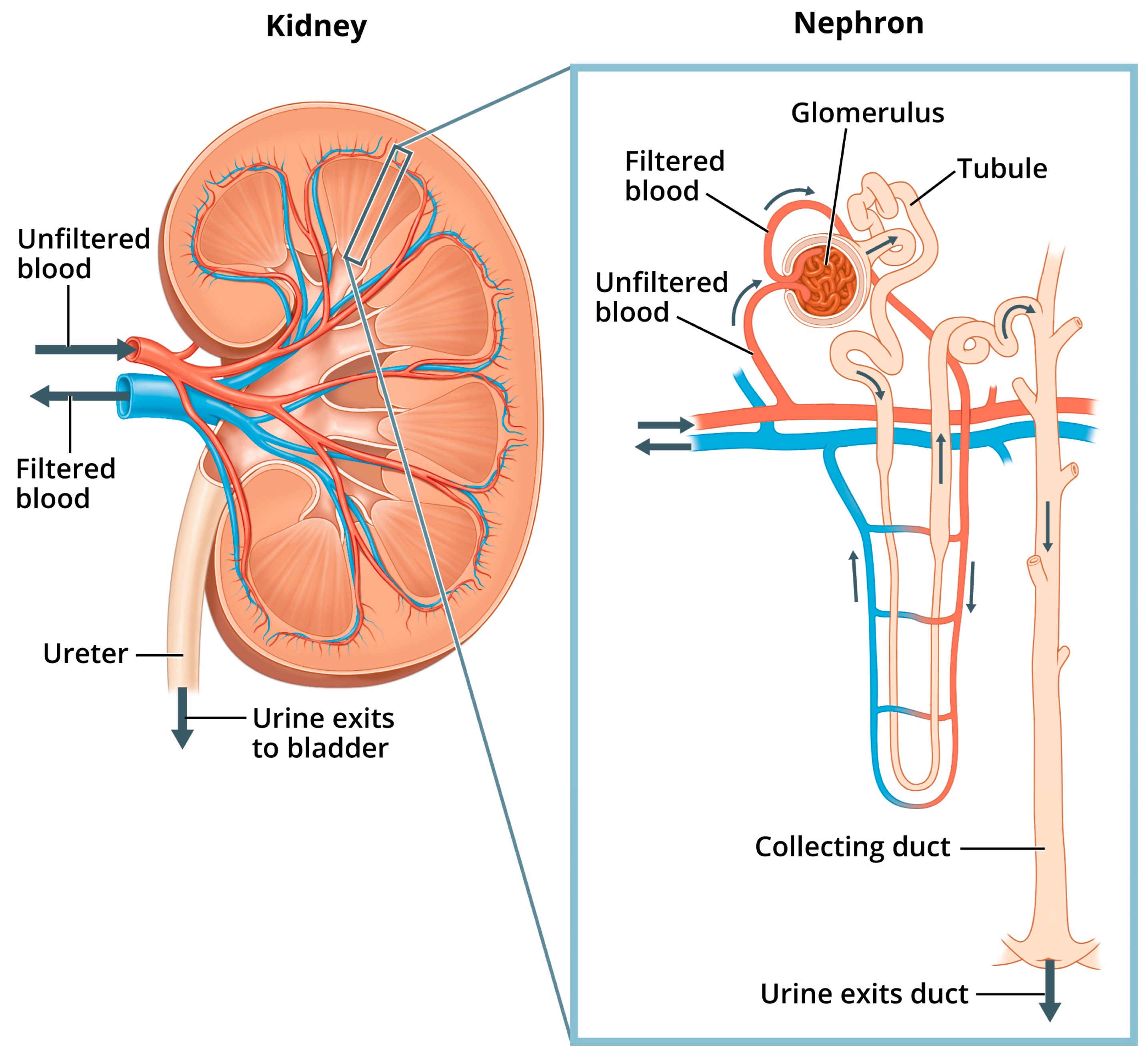
Nephron Regulation of Pi
4. Renal Pathologies and Cancer
4.1. Renal Tubular Dysfunction
4.2. Glomerular Dysfunction
4.3. Kidney Stone Formation
4.4. Polycystic Kidney Disease
4.5. Renal Fibrosis
5. Fibroblast Growth Factor Receptors and Pi in Cancer
6. Pi Cell Transport and Cell Signaling in Cancer
7. Pi, RNA, and the Tumor Microenvironment
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jordan, B.R. The Hiroshima/Nagasaki Survivor Studies: Discrepancies Between Results and General Perception. Genetics 2016, 203, 1505–1512. [Google Scholar] [CrossRef]
- Radiation Effects Research Foundation. About RERF. Available online: https://www.rerf.or.jp/en/about/ (accessed on 16 January 2025).
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; International Agency for Research on Cancer: Lyon, France, 1993; Volume 56. [Google Scholar]
- IARC. List of Classifications by Cancer Sites with Sufficient or Limited Evidence in Humans, IARC Monographs Volumes 1–138a. Available online: https://monographs.iarc.who.int/wp-content/uploads/2019/07/Classifications_by_cancer_site.pdf (accessed on 7 June 2025).
- American Cancer Society. Known and Probable Human Carcinogens. Available online: https://www.cancer.org/cancer/risk-prevention/understanding-cancer-risk/known-and-probable-human-carcinogens.html (accessed on 1 February 2025).
- National Cancer Institute. The Genetics of Cancer. Available online: https://www.cancer.gov/about-cancer/causes-prevention/genetics (accessed on 1 February 2025).
- DeMarini, D.M. The role of genotoxicity in carcinogenesis. In Tumour Site Concordance and Mechanisms of Carcinogenesis; Baan, R.A., Stewart, B.W., Straif, K., Eds.; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Moon, J.; Kitty, I.; Renata, K.; Qin, S.; Zhao, F.; Kim, W. DNA Damage and Its Role in Cancer Therapeutics. Int. J. Mol. Sci. 2023, 24, 4741. [Google Scholar] [CrossRef]
- Paytubi, S.; Benavente, Y.; Montoliu, A.; Binefa, G.; Brotons, M.; Ibáñez, R.; Ochoa, C.; Peremiquel-Trillas, P.; Serrano, B.; Travier, N.; et al. Everything causes cancer? Beliefs and attitudes towards cancer prevention among anti-vaxxers, flat earthers, and reptilian conspiracists: Online cross sectional survey. Br. Med. J. 2022, 379, e072561. [Google Scholar] [CrossRef]
- National Human Genome Research Institute. Carcinogen. Available online: https://www.genome.gov/genetics-glossary/Carcinogen (accessed on 1 February 2025).
- Cohen, E.P.; Robbins, M.E. Radiation nephropathy. Semin. Nephrol. 2003, 23, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Uber, A.M.; Sutherland, S.M. Nephrotoxins and nephrotoxic acute kidney injury. Pediatr. Nephrol. 2020, 35, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, G.E. Toxic nephropathy: Adverse renal effects caused by drugs and chemicals. J. Am. Med. Assoc. 1965, 191, 849–850. [Google Scholar] [CrossRef]
- Jagieła, J.; Bartnicki, P.; Rysz, J. Nephrotoxicity as a Complication of Chemotherapy and Immunotherapy in the Treatment of Colorectal Cancer, Melanoma and Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 4618. [Google Scholar] [CrossRef]
- Ding, J.; Etzel, R.A. Environmental Nephrotoxins. In Pediatric Kidney Disease; Springer: Berlin/Heidelberg, Germany, 2023; pp. 2019–2037. [Google Scholar]
- Environment and Climate Change Canada—Health Canada. State of Per- and Polyfluoroalkyl Substances (PFAS) Report. Available online: https://www.canada.ca/en/environment-climate-change/services/evaluating-existing-substances/state-per-polyfluoroalkyl-substances-report.html (accessed on 7 March 2025).
- Fu, Y.C.; Xu, Z.L.; Zhao, M.Y.; Xu, K. The Association Between Smoking and Renal Function in People Over 20 Years Old. Front. Med. 2022, 9, 870278. [Google Scholar] [CrossRef]
- Lang, S.M.; Schiffl, H. Smoking status, cadmium, and chronic kidney disease. Ren. Replace. Ther. 2024, 10, 17. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshihisa, A.; Maki, T.; Takeishi, Y. Effects of daily alcohol intake on glomerular filtration rate over three years. Fukushima J. Med. Sci. 2021, 67, 1–7. [Google Scholar] [CrossRef]
- Epstein, M. Alcohol’s impact on kidney function. Alcohol. Health Res. World 1997, 21, 84–92. [Google Scholar] [PubMed]
- National Cancer Institute. Alcohol and Cancer Risk. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/alcohol/alcohol-fact-sheet (accessed on 22 February 2025).
- National Cancer Institute. Tobacco. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/tobacco (accessed on 22 February 2025).
- Szymczak, A.; Kusztal, M.; Krajewska, M. Overhydration: A cause or an effect of kidney damage and how to treat it. Adv. Clin. Exp. Med. 2021, 30, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Healthline. What You Need to Know About Sodium If You Have Chronic Kidney Disease (CKD). Available online: https://www.healthline.com/health/kidney-disease/chronic-kidney-disease-sodium (accessed on 11 April 2025).
- Rubio-Aliaga, I.; Krapf, R. Phosphate intake, hyperphosphatemia, and kidney function. Pflug. Arch. 2022, 474, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P. Paracelsus Revisited: The Dose Concept in a Complex World. Basic. Clin. Pharmacol. Toxicol. 2016, 119, 126–132. [Google Scholar] [CrossRef]
- IARC Scientific Publications. Tumour Site Concordance and Mechanisms of Carcinogenesis. Available online: https://www.ncbi.nlm.nih.gov/books/NBK570327/ (accessed on 18 January 2025).
- Kaufman, D.G. Assessment of Carcinogenicity: Generic Issues and Their Application to Diesel Exhaust. Available online: https://www.ncbi.nlm.nih.gov/books/NBK218143/ (accessed on 18 January 2025).
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small molecule metabolites: Discovery of biomarkers and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef]
- Ruan, Y.; Fang, X.; Guo, T.; Liu, Y.; Hu, Y.; Wang, X.; Hu, Y.; Gao, L.; Li, Y.; Pi, J.; et al. Metabolic reprogramming in the arsenic carcinogenesis. Ecotoxicol. Environ. Saf. 2022, 229, 113098. [Google Scholar] [CrossRef]
- Arnst, J.L.; Beck, G.R., Jr. Modulating phosphate consumption, a novel therapeutic approach for the control of cancer cell proliferation and tumorigenesis. Biochem. Pharmacol. 2021, 183, 114305. [Google Scholar] [CrossRef]
- Brown, R.B.; Bigelow, P.; Dubin, J.A.; Mielke, J.G. High Dietary Phosphorus Is Associated with Increased Breast Cancer Risk in a U.S. Cohort of Middle-Aged Women. Nutrients 2023, 15, 3735. [Google Scholar] [CrossRef]
- Brown, R.B.; Mielke, J.G. Modulating Factors in Cancer: Dietary Phosphorus. In Handbook of Public Health Nutrition: International, National, and Regional Perspectives; Preedy, V.R., Patel, V.B., Eds.; Springer Nature: Cham, Switzerland, 2025; pp. 1–18. [Google Scholar]
- Meraz-Munoz, A.; Langote, A.; Jhaveri, K.D.; Izzedine, H.; Gudsoorkar, P. Acute Kidney Injury in the Patient with Cancer. Diagnostics 2021, 11, 611. [Google Scholar] [CrossRef]
- Lees, J.S.; Elyan, B.M.P.; Herrmann, S.M.; Lang, N.N.; Jones, R.J.; Mark, P.B. The ‘other’ big complication: How chronic kidney disease impacts on cancer risks and outcomes. Nephrol. Dial. Transplant. 2023, 38, 1071–1079. [Google Scholar] [CrossRef]
- Rosner, M.H.; Jhaveri, K.D.; McMahon, B.A.; Perazella, M.A. Onconephrology: The intersections between the kidney and cancer. CA A Cancer J. Clin. 2021, 71, 47–77. [Google Scholar] [CrossRef]
- Brown, R.B.; Razzaque, M.S. Dysregulation of phosphate metabolism and conditions associated with phosphate toxicity. BoneKEy Rep. 2015, 4, 705. [Google Scholar] [CrossRef]
- Wolfswinkel, J.F.; Furtmueller, E.; Wilderom, C.P.M. Using grounded theory as a method for rigorously reviewing literature. Eur. J. Inf. Syst. 2013, 22, 45–55. [Google Scholar] [CrossRef]
- Yazdanpanah, N.; Dochy, F.; Darmstadt, G.L.; Peters, G.J.; Tsitlakidis, A.; Aifantis, E.C.; Cerda, A.; Comini, E.; Brand, S.; Gupta, M.; et al. Cancer: A Complex Problem Requiring Interdisciplinary Research. In Cancer Treatment: An Interdisciplinary Approach; Rezaei, N., Ed.; Springer Nature: Cham, Switzerland, 2023; pp. 1–45. [Google Scholar]
- Barnett, L.M.A.; Cummings, B.S. Nephrotoxicity and Renal Pathophysiology: A Contemporary Perspective. Toxicol. Sci. 2018, 164, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.; Frain, H.; Ryan, M.P.; Slattery, C.; McMorrow, T. Mechanisms of chemical carcinogenesis in the kidneys. Int. J. Mol. Sci. 2013, 14, 19416–19433. [Google Scholar] [CrossRef] [PubMed]
- Knox, F.G.; Schneider, E.G.; Willis, L.R.; Strandhoy, J.W.; Ott, C.E. Editorial: Site and control of phosphate reabsorption by the kidney. Kidney Int. 1973, 3, 347–353. [Google Scholar] [CrossRef]
- Blaine, J.; Chonchol, M.; Levi, M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 1257–1272. [Google Scholar] [CrossRef]
- Kaufman, D.P.; Basit, H.; Knohl, S.J. Physiology, Glomerular Filtration Rate; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Rout, P.; Jialal, I. Hyperphosphatemia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Voormolen, N.; Noordzij, M.; Grootendorst, D.C.; Beetz, I.; Sijpkens, Y.W.; van Manen, J.G.; Boeschoten, E.W.; Huisman, R.M.; Krediet, R.T.; Dekker, F.W.; et al. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol. Dial. Transplant. 2007, 22, 2909–2916. [Google Scholar] [CrossRef]
- Hashmi, M.F.; Benjamin, O.; Lappin, S.L. End-Stage Renal Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Portale, A.A.; Halloran, B.P.; Morris, R.C., Jr. Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D. J. Clin. Investig. 1987, 80, 1147–1154. [Google Scholar] [CrossRef]
- Calvo, M.S.; Moshfegh, A.J.; Tucker, K.L. Assessing the Health Impact of Phosphorus in the Food Supply: Issues and Considerations. Adv. Nutr. 2014, 5, 104–113. [Google Scholar] [CrossRef]
- Brown, R.B.; Razzaque, M.S. Phosphate toxicity and tumorigenesis. Biochim. et Biophys. Acta (BBA)-Rev. Cancer 2018, 1869, 303–309. [Google Scholar] [CrossRef]
- Saly, D.L.; Eswarappa, M.S.; Street, S.E.; Deshpande, P. Renal Cell Cancer and Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2021, 28, 460–468.e461. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, S.R.; Yarrarapu, S.N.S.; Aeddula, N.R. Nephrocalcinosis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2025. [Google Scholar]
- Liang, X.; Li, Y.; Wang, P.; Liu, H. Key regulators of vascular calcification in chronic kidney disease: Hyperphosphatemia, BMP2, and RUNX2. PeerJ 2024, 12, e18063. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.W.; Adler, S.; Budoff, M.; Takasu, J.; Ashai, J.; Mehrotra, R. Prevalence and prognostic significance of renal artery calcification in patients with diabetes and proteinuria. Clin. J. Am. Soc. Nephrol. 2010, 5, 2093–2100. [Google Scholar] [CrossRef]
- Rifkin, D.E.; Ix, J.H.; Wassel, C.L.; Criqui, M.H.; Allison, M.A. Renal artery calcification and mortality among clinically asymptomatic adults. J. Am. Coll. Cardiol. 2012, 60, 1079–1085. [Google Scholar] [CrossRef]
- Ding, M.; Zhang, Q.; Zhang, M.; Jiang, X.; Wang, M.; Ni, L.; Gong, W.; Huang, B.; Chen, J. Phosphate Overload Stimulates Inflammatory Reaction via PiT-1 and Induces Vascular Calcification in Uremia. J. Ren. Nutr. 2022, 32, 178–188. [Google Scholar] [CrossRef]
- Fujimura, R.; Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Namba-Hamano, T.; Minami, S.; Sakai, S.; Matsuda, J.; Hesaka, A.; Yonishi, H.; et al. Autophagy protects kidney from phosphate-induced mitochondrial injury. Biochem. Biophys. Res. Commun. 2020, 524, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Amanzadeh, J.; Reilly, R.F. Hypophosphatemia: An evidence-based approach to its clinical consequences and management. Nat. Clin. Pract. Nephrol. 2006, 2, 136–148. [Google Scholar] [CrossRef]
- Adhikari, S.; Mamlouk, O.; Rondon-Berrios, H.; Workeneh, B.T. Hypophosphatemia in cancer patients. Clin. Kidney J. 2021, 14, 2304–2315. [Google Scholar] [CrossRef]
- Dukmak, O.N.; Ayyad, M.; Albandak, M.; Hamadah, A.; Gharaibeh, K. Tumor Genesis Syndrome Presenting as Severe Hypophosphatemia in a Patient With T-Cell Acute Lymphoblastic Leukemia. Cureus 2023, 15, e38815. [Google Scholar] [CrossRef]
- Osuka, S.; Razzaque, M.S. Can features of phosphate toxicity appear in normophosphatemia? J. Bone Miner. Metab. 2012, 30, 10–18. [Google Scholar] [CrossRef]
- Al-Naimi, M.S.; Rasheed, H.A.; Hussien, N.R.; Al-Kuraishy, H.M.; Al-Gareeb, A.I. Nephrotoxicity: Role and significance of renal biomarkers in the early detection of acute renal injury. J. Adv. Pharm. Technol. Res. 2019, 10, 95–99. [Google Scholar] [CrossRef]
- Nogawa, K.; Watanabe, Y.; Sakuma, S.; Sakurai, M.; Nishijo, M.; Ishizaki, M.; Morikawa, Y.; Kido, T.; Nakagawa, H.; Suwazono, Y. Renal tubular dysfunction and cancer mortality in the Japanese general population living in cadmium-non-contaminated areas. J. Appl. Toxicol. 2022, 42, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Hodgkins, K.S.; Schnaper, H.W. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol. 2012, 27, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Lumlertgul, D.; Burke, T.J.; Gillum, D.M.; Alfrey, A.C.; Harris, D.C.; Hammond, W.S.; Schrier, R.W. Phosphate depletion arrests progression of chronic renal failure independent of protein intake. Kidney Int. 1986, 29, 658–666. [Google Scholar] [CrossRef]
- Wang, P.; Chen, B.; Huang, Y.; Li, J.; Cao, D.; Chen, Z.; Li, J.; Ran, B.; Yang, J.; Wang, R.; et al. The relationship between nonsteroidal anti-inflammatory drugs and cancer incidence: An umbrella review. Heliyon 2024, 10, e23203. [Google Scholar] [CrossRef]
- Ha, Y.S.; Tchey, D.U.; Kang, H.W.; Kim, Y.J.; Yun, S.J.; Lee, S.C.; Kim, W.J. Phosphaturia as a promising predictor of recurrent stone formation in patients with urolithiasis. Korean J. Urol. 2010, 51, 54–59. [Google Scholar] [CrossRef]
- Taylor, E.N.; Hoofnagle, A.N.; Curhan, G.C. Calcium and phosphorus regulatory hormones and risk of incident symptomatic kidney stones. Clin. J. Am. Soc. Nephrol. 2015, 10, 667–675. [Google Scholar] [CrossRef]
- Negri, A.L.; Spivacow, R.; Del Valle, E.; Fradinger, E.; Marino, A.; Zanchetta, J.R. Renal phosphate leak in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Urol. Res. 2003, 31, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Halter, T.J.; Borah, B.M.; Nancollas, G.H. Aggregation of Calcium Phosphate and Oxalate Phases in the Formation of Renal Stones. Cryst. Growth Des. 2015, 15, 204–211. [Google Scholar] [CrossRef]
- van de Pol, J.A.A.; van den Brandt, P.A.; Schouten, L.J. Kidney stones and the risk of renal cell carcinoma and upper tract urothelial carcinoma: The Netherlands Cohort Study. Br. J. Cancer 2019, 120, 368–374. [Google Scholar] [CrossRef]
- Omede, F.; Zhang, S.; Johnson, C.; Daniel, E.; Zhang, Y.; Fields, T.A.; Boulanger, J.; Liu, S.; Ahmed, I.; Umar, S.; et al. Dietary phosphate restriction attenuates polycystic kidney disease in mice. Am. J. Physiol.-Ren. Physiol. 2020, 318, F35–F42. [Google Scholar] [CrossRef]
- Nitta, K.; Kataoka, H.; Manabe, S.; Makabe, S.; Akihisa, T.; Ushio, Y.; Seki, M.; Tsuchiya, K.; Hoshino, J.; Mochizuki, T. Association of hyperphosphatemia with renal prognosis in patients with autosomal dominant polycystic kidney disease. Clin. Exp. Nephrol. 2025, 29, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.-W.; Shih, C.-M.; Li, S.-Y.; Tseng, S.-H.; Dubey, R.; Wu, M.-S. Susceptibility of Developing Renal and Lung Cancer in Polycystic Kidney Disease Patients: An Evidence in Reaching Consensus. Eur. J. Cancer Care 2023, 2023, 5036299. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef]
- Shen, Z.-J.; Hu, J.; Shiizaki, K.; Kuro-o, M.; Malter, J.S. Phosphate–Induced Renal Fibrosis Requires the Prolyl Isomerase Pin1. PLoS ONE 2016, 11, e0150093. [Google Scholar] [CrossRef]
- Chen, J.Y.; Yiu, W.H.; Tang, P.M.; Tang, S.C. New insights into fibrotic signaling in renal cell carcinoma. Front. Cell Dev. Biol. 2023, 11, 1056964. [Google Scholar] [CrossRef]
- Lewis, E.; Seltun, F.; Razzaque, M.S.; He, P. Phosphate Toxicity and Epithelial to Mesenchymal Transition. Adv. Exp. Med. Biol. 2022, 1362, 73–84. [Google Scholar] [CrossRef]
- Ewendt, F.; Feger, M.; Föller, M. Role of Fibroblast Growth Factor 23 (FGF23) and αKlotho in Cancer. Front. Cell Dev. Biol. 2020, 8, 601006. [Google Scholar] [CrossRef]
- Lee, E.K.; Martinez, M.C.; Blakely, K.; Santos, K.D.; Hoang, V.C.; Chow, A.; Emmenegger, U. FGF23: Mediator of poor prognosis in a sizeable subgroup of patients with castration-resistant prostate cancer presenting with severe hypophosphatemia? Med. Hypotheses 2014, 83, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Cekin, R.; Arici, S.; Atci, M.M.; Secmeler, S.; Cihan, S. The clinical importance of fibroblast growth factor 23 on breast cancer patients. J. Med. Investig. 2020, 4, 471–476. [Google Scholar]
- Tebben, P.J.; Kalli, K.R.; Cliby, W.A.; Hartmann, L.C.; Grande, J.P.; Singh, R.J.; Kumar, R. Elevated fibroblast growth factor 23 in women with malignant ovarian tumors. Mayo Clin. Proc. 2005, 80, 745–751. [Google Scholar] [CrossRef]
- Jacobs, E.; Martinez, M.E.; Buckmeier, J.; Lance, P.; May, M.; Jurutka, P. Circulating fibroblast growth factor-23 is associated with increased risk for metachronous colorectal adenoma. J. Carcinog. 2011, 10, 3. [Google Scholar] [CrossRef]
- Yang, L.; Cai, Y.; Wang, Y.; Huang, Y.; Zhang, C.; Ma, H.; Zhou, J.-G. Fibroblast Growth Factor 23 is a Potential Prognostic Biomarker in Uterine Sarcoma. Technol. Cancer Res. Treat. 2024, 23, 15330338241245924. [Google Scholar] [CrossRef]
- Zhang, P.; Yue, L.; Leng, Q.; Chang, C.; Gan, C.; Ye, T.; Cao, D. Targeting FGFR for cancer therapy. J. Hematol. Oncol. 2024, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Xiao, Z.; Smith, J.C.; Quarles, L.D. Structural asymmetry in FGF23 signaling. Trends Pharmacol. Sci. 2023, 44, 862–864. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Wang, Z.; Anderson, K.S. Therapeutic Targeting of FGFR Signaling in Head and Neck Cancer. Cancer J. 2022, 28, 354–362. [Google Scholar] [CrossRef]
- Tsimafeyeu, I.; Bratslavsky, G. Fibroblast growth factor receptor 1 as a target for the therapy of renal cell carcinoma. Oncology 2015, 88, 321–331. [Google Scholar] [CrossRef]
- Fan, S.; Chen, Y.; Wang, W.; Xu, W.; Tian, M.; Liu, Y.; Zhou, Y.; Liu, D.; Xia, Q.; Dong, L. Pharmacological and Biological Targeting of FGFR1 in Cancer. Curr. Issues Mol. Biol. 2024, 46, 13131–13150. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Ózsvári, B.; Sotgia, F.; Lisanti, M.P. High ATP Production Fuels Cancer Drug Resistance and Metastasis: Implications for Mitochondrial ATP Depletion Therapy. Front. Oncol. 2021, 11, 740720. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the Origin of ATP Synthesis in Cancer. iScience 2020, 23, 101761. [Google Scholar] [CrossRef]
- Ancey, P.-B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Barreca, F.; Aventaggiato, M.; Vitiello, L.; Sansone, L.; Russo, M.A.; Mai, A.; Valente, S.; Tafani, M. SIRT5 Activation and Inorganic Phosphate Binding Reduce Cancer Cell Vitality by Modulating Autophagy/Mitophagy and ROS. Antioxidants 2023, 12, 1635. [Google Scholar] [CrossRef]
- Bi, Q.-C.; Luo, R.-G.; Li, Y.-S.; Zhao, J.; Fu, X.; Chen, H.; Lv, Y.-F.; Liu, Z.-X.; Liang, Q.-R.; Tang, Q. Low Inorganic Phosphate Stress Inhibits Liver Cancer Progression: From In Vivo to In Vitro. Adv. Ther. 2022, 5, 2100224. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, X.; Zhang, L.; Yang, F.; Qin, L.; Zhang, D.; Qin, Y. SLC34A2 regulates miR-25-Gsk3β signaling pathway to affect tumor progression in gastric cancer stem cell-like cells. Mol. Carcinog. 2018, 57, 440–450. [Google Scholar] [CrossRef]
- Jiang, Z.; Hao, Y.; Ding, X.; Zhang, Z.; Liu, P.; Wei, X.; Xi, J. The effects and mechanisms of SLC34A2 on tumorigenicity in human non-small cell lung cancer stem cells. Tumour Biol. 2016, 37, 10383–10392. [Google Scholar] [CrossRef]
- Liu, L.; Yang, Y.; Zhou, X.; Yan, X.; Wu, Z. Solute carrier family 34 member 2 overexpression contributes to tumor growth and poor patient survival in colorectal cancer. Biomed. Pharmacother. 2018, 99, 645–654. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, J.; Yu, X.; Wu, Q.; Cao, H.; Dai, X.; Chen, H. SLC34A2 promotes cancer proliferation and cell cycle progression by targeting TMPRSS3 in colorectal cancer. Pathol. Res. Pr. 2022, 229, 153706. [Google Scholar] [CrossRef]
- Shyian, M.; Gryshkova, V.; Kostianets, O.; Gorshkov, V.; Gogolev, Y.; Goncharuk, I.; Nespryadko, S.; Vorobjova, L.; Filonenko, V.; Kiyamova, R. Quantitative analysis of SLC34A2 expression in different types of ovarian tumors. Exp. Oncol. 2011, 33, 94–98. [Google Scholar] [PubMed]
- Vlasenkova, R.; Nurgalieva, A.; Akberova, N.; Bogdanov, M.; Kiyamova, R. Characterization of SLC34A2 as a Potential Prognostic Marker of Oncological Diseases. Biomolecules 2021, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Chen, C.; Gao, Y.; Zheng, Z.-S.; Xu, Y.; Yun, M.; Weng, H.-W.; Xie, D.; Ye, S.; Zhang, J.-X. Overexpression of SLC34A2 is an independent prognostic indicator in bladder cancer and its depletion suppresses tumor growth via decreasing c-Myc expression and transcriptional activity. Cell Death Dis. 2017, 8, e2581. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, Z.; Xu, Y.; Zhao, L.; Zhang, P.; Gao, H.; Wang, Q.; Xia, Q. Low expression of SLC34A1 is associated with poor prognosis in clear cell renal cell carcinoma. BMC Urol. 2023, 23, 45. [Google Scholar] [CrossRef]
- Carpenter, T.O.; Bergwitz, C.; Insogna, K.L. Chapter 20—Phosphorus homeostasis and related disorders. In Principles of Bone Biology, 4th ed.; Bilezikian, J.P., Martin, T.J., Clemens, T.L., Rosen, C.J., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 469–507. [Google Scholar]
- Clerin, V.; Saito, H.; Filipski, K.J.; Nguyen, A.H.; Garren, J.; Kisucka, J.; Reyes, M.; Jüppner, H. Selective pharmacological inhibition of the sodium-dependent phosphate cotransporter NPT2a promotes phosphate excretion. J. Clin. Investig. 2020, 130, 6510–6522. [Google Scholar] [CrossRef]
- He, J.; Zhou, M.; Li, X.; Gu, S.; Cao, Y.; Xing, T.; Chen, W.; Chu, C.; Gu, F.; Zhou, J.; et al. SLC34A2 simultaneously promotes papillary thyroid carcinoma growth and invasion through distinct mechanisms. Oncogene 2020, 39, 2658–2675. [Google Scholar] [CrossRef]
- Jin, H.; Xu, C.-X.; Lim, H.-T.; Park, S.-J.; Shin, J.-Y.; Chung, Y.-S.; Park, S.-C.; Chang, S.-H.; Youn, H.-J.; Lee, K.-H. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am. J. Respir. Crit. Care Med. 2009, 179, 59–68. [Google Scholar] [CrossRef]
- Turdo, A.; D’Accardo, C.; Glaviano, A.; Porcelli, G.; Colarossi, C.; Colarossi, L.; Mare, M.; Faldetta, N.; Modica, C.; Pistone, G.; et al. Targeting Phosphatases and Kinases: How to Checkmate Cancer. Front. Cell Dev. Biol. 2021, 9, 690306. [Google Scholar] [CrossRef]
- Banerjee, S.; Drapkin, R.; Richardson, D.L.; Birrer, M. Targeting NaPi2b in ovarian cancer. Cancer Treat. Rev. 2023, 112, 102489. [Google Scholar] [CrossRef]
- Hong, S.H.; Minai-Tehrani, A.; Chang, S.H.; Jiang, H.L.; Lee, S.; Lee, A.Y.; Seo, H.W.; Chae, C.; Beck, G.R., Jr.; Cho, M.H. Knockdown of the sodium-dependent phosphate co-transporter 2b (NPT2b) suppresses lung tumorigenesis. PLoS ONE 2013, 8, e77121. [Google Scholar] [CrossRef]
- Russo-Abrahão, T.; Lacerda-Abreu, M.A.; Gomes, T.; Cosentino-Gomes, D.; Carvalho-de-Araújo, A.D.; Rodrigues, M.F.; Oliveira, A.C.L.; Rumjanek, F.D.; Monteiro, R.Q.; Meyer-Fernandes, J.R. Characterization of inorganic phosphate transport in the triple-negative breast cancer cell line, MDA-MB-231. PLoS ONE 2018, 13, e0191270. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Cosentino-Gomes, D.; Nascimento, M.T.C.; Carvalho-Kelly, L.F.; Gomes, T.; Rodrigues, M.F.; König, S.; Rumjanek, F.D.; Monteiro, R.Q.; et al. H(+)-dependent inorganic phosphate transporter in breast cancer cells: Possible functions in the tumor microenvironment. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Worsley, C.M.; Veale, R.B.; Mayne, E.S. The acidic tumour microenvironment: Manipulating the immune response to elicit escape. Hum. Immunol. 2022, 83, 399–408. [Google Scholar] [CrossRef]
- Bansal, V.K. Serum inorganic phosphorus. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Horowitz, A.; Chanez-Paredes, S.D.; Haest, X.; Turner, J.R. Paracellular permeability and tight junction regulation in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Chiruvella, V.; Annamaraju, P.; Guddati, A.K. Management of nephrotoxicity of chemotherapy and targeted agents: 2020. Am. J. Cancer Res. 2020, 10, 4151–4164. [Google Scholar]
- Tang, J.; Yang, N.; Pan, S.; Ren, P.; Chen, M.; Jin, J.; He, Q.; Zeng, Y. The renal damage and mechanisms relevant to antitumoral drugs. Front. Oncol. 2023, 13, 1331671. [Google Scholar] [CrossRef]
- American Cancer Society. Second Cancers Related to Treatment. Available online: https://www.cancer.org/cancer/survivorship/long-term-health-concerns/second-cancers-in-adults/treatment-risks.html (accessed on 16 February 2025).
- Chen, T.; Fallah, M.; Sundquist, K.; Liu, H.; Hemminki, K. Risk of subsequent cancers in renal cell carcinoma survivors with a family history. Eur. J. Cancer 2014, 50, 2108–2118. [Google Scholar] [CrossRef]
- Casey, S.C.; Vaccari, M.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Barcellos-Hoff, M.H.; Brown, D.G.; Chapellier, M.; Christopher, J.; Curran, C.S.; et al. The effect of environmental chemicals on the tumor microenvironment. Carcinogenesis 2015, 36 (Suppl. S1), S160–S183. [Google Scholar] [CrossRef]
- Wu, D.J. Oversupply of limiting cell resources and the evolution of cancer cells: A review. Front. Ecol. Evol. 2021, 9, 653622. [Google Scholar] [CrossRef]
- Isanta-Navarro, J.; Prater, C.; Peoples, L.M.; Loladze, I.; Phan, T.; Jeyasingh, P.D.; Church, M.J.; Kuang, Y.; Elser, J.J. Revisiting the growth rate hypothesis: Towards a holistic stoichiometric understanding of growth. Ecol. Lett. 2022, 25, 2324–2339. [Google Scholar] [CrossRef]
- National Human Genome Research Institute. Phosphate backbone. Available online: https://www.genome.gov/genetics-glossary/Phosphate-Backbone (accessed on 7 March 2025).
- Nature Education. Ribosomes, Transcription, and Translation. Available online: https://www.nature.com/scitable/topicpage/ribosomes-transcription-and-translation-14120660/ (accessed on 20 February 2025).
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef]
- Bahadori, M. New Insights into Connection of Nucleolar Functions and Cancer. Tanaffos 2019, 18, 173–179. [Google Scholar] [PubMed]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome Biogenesis: A Central Player in Cancer Metastasis and Therapeutic Resistance. Cancer Res. 2022, 82, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.N.; Griffin, A.C. Phosphorus incorporation into nucleic acids and proteins of liver nuclei of normal and azo dye-fed rats. Cancer Res. 1955, 15, 456–461. [Google Scholar]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Bobko, A.A.; Eubank, T.D.; Driesschaert, B.; Dhimitruka, I.; Evans, J.; Mohammad, R.; Tchekneva, E.E.; Dikov, M.M.; Khramtsov, V.V. Interstitial inorganic phosphate as a tumor microenvironment marker for tumor progression. Sci. Rep. 2017, 7, 41233. [Google Scholar] [CrossRef]
- Eubank, T.D.; Bobko, A.A.; Hoblitzell, E.H.; Gencheva, M.; Driesschaert, B.; Khramtsov, V.V. In Vivo Electron Paramagnetic Resonance Molecular Profiling of Tumor Microenvironment upon Tumor Progression to Malignancy in an Animal Model of Breast Cancer. Mol. Imaging Biol. 2023, 26, 424–434. [Google Scholar] [CrossRef]
- Lin, Y.; McKinnon, K.E.; Ha, S.W.; Beck, G.R., Jr. Inorganic phosphate induces cancer cell mediated angiogenesis dependent on forkhead box protein C2 (FOXC2) regulated osteopontin expression. Mol. Carcinog. 2015, 54, 926–934. [Google Scholar] [CrossRef]
- Kourie, C.M.; Ghamlouche, F.; Hachem, S.; Yehya, A.; Jaafar, L.; El-Mallah, C.; Abou-Kheir, W.; Obeid, O. Effect of Inorganic Phosphorus Manipulation on the Growth and Progression of Prostate Cancer Cells In Vitro. Int. J. Mol. Sci. 2025, 26, 4762. [Google Scholar] [CrossRef]
- Scott, R.P.; Quaggin, S.E. The cell biology of renal filtration. J. Cell Biol. 2015, 209, 199–210. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Nephrotoxin (Cancer Site) | |
|---|---|
| Ammonia | Methyl parathion |
| Arsenic (kidney, urinary bladder, liver, bile duct, lung, skin, prostate) | Mercury |
| Aristolochic acid (renal pelvis and ureter) | Naphthalene |
| Barium | Ochratoxin A |
| Cadmium (kidney, lung, prostate) | Pentachlorophenol (lymphoma and multiple myeloma) |
| Carbon tetrachloride | Per- and Polyfluoroalkylated substances |
| Chloromethane (bile duct and lymphoma) | Thallium |
| Chromate and chromium (VI) (lung, nasal cavity and paranasal sinus) | Trichloroethylene (liver, bile duct, kidney, lymphoma) |
| Copper sulfate | Uranium (lung) |
| Fluoride | 1,2 dibromomethane |
| Formaldehyde (nasal cavity and paranasal sinus, acute and chronic myeloid leukemias, other acute non-lymphocytic leukemia) | 1,2 dichloromethane (lymphoma, bile duct) |
| Lead (stomach) | 1,2 dichloropropane (bile duct) |
| Melamine |
| Chronic Kidney Disease | Glomerular Filtration Rate |
|---|---|
| Stage 1—normal | >90 mL/min |
| Stage 2—mild | 60 to 89 mL/min |
| Stage 3a—mild to moderate | 45 to 59 mL/min |
| Stage 3b—moderate to severe | 30 to 44 mL/min |
| Stage 4—severe | 15 to 29 mL/min |
| Stage 5—failure | <15 mL/min |
| Coding RNA Type | Description |
|---|---|
| Messenger RNA | “Messenger RNA (mRNA) molecules carry the coding sequences for protein synthesis and are called transcripts”. |
| Ribosomal RNA | “Ribosomal RNA (rRNA) molecules form the core of a cell’s ribosomes (the structures in which protein synthesis takes place)”. |
| Transfer RNA | ‘Transfer RNA (tRNA) molecules carry amino acids to the ribosomes during protein synthesis”. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, R.B.; Mielke, J.G. Carcinogenesis Associated with Toxin Nephropathy: Proposed Mediation by Phosphate Toxicity. Cells 2025, 14, 952. https://doi.org/10.3390/cells14130952
Brown RB, Mielke JG. Carcinogenesis Associated with Toxin Nephropathy: Proposed Mediation by Phosphate Toxicity. Cells. 2025; 14(13):952. https://doi.org/10.3390/cells14130952
Chicago/Turabian StyleBrown, Ronald B., and John G. Mielke. 2025. "Carcinogenesis Associated with Toxin Nephropathy: Proposed Mediation by Phosphate Toxicity" Cells 14, no. 13: 952. https://doi.org/10.3390/cells14130952
APA StyleBrown, R. B., & Mielke, J. G. (2025). Carcinogenesis Associated with Toxin Nephropathy: Proposed Mediation by Phosphate Toxicity. Cells, 14(13), 952. https://doi.org/10.3390/cells14130952