
Yoda1 Inhibits TGFβ-Induced Cardiac Fibroblast Activation via a BRD4-Dependent Pathway


Abstract
1. Introduction
2. Materials and Methods
2.1. 3T3 Cell Culture and Yoda1 Treatment
2.2. Treatment of 3T3 Cells with Ion Channel Modulators and Pharmacological Inhibitors
2.3. Murine Cardiac Fibroblast Isolation and Culture
2.4. Treatment of Murine Primary Cardiac Fibroblasts
2.5. RNA Isolation
2.6. Transcriptomic Sequencing
2.7. siRNA Transfection
2.8. RT-PCR Analysis
2.9. BRD4 Overexpressing Adenoviral Vector Production and Cell Transduction
3. Statistical Analysis
4. Results
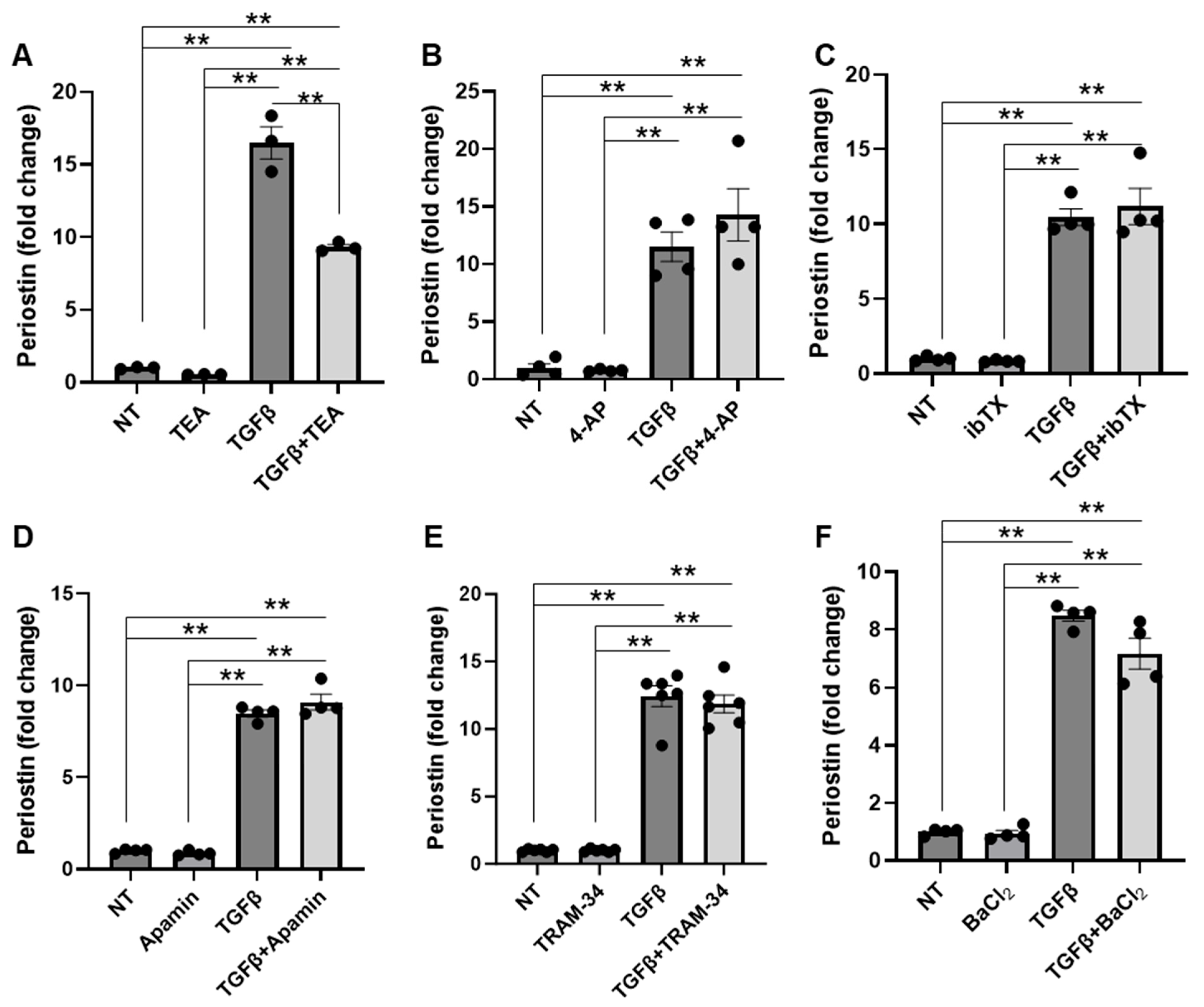
4.1. Potassium Channels Do Not Significantly Contribute to TGFβ-Induced NIH/3T3 Fibroblast Activation
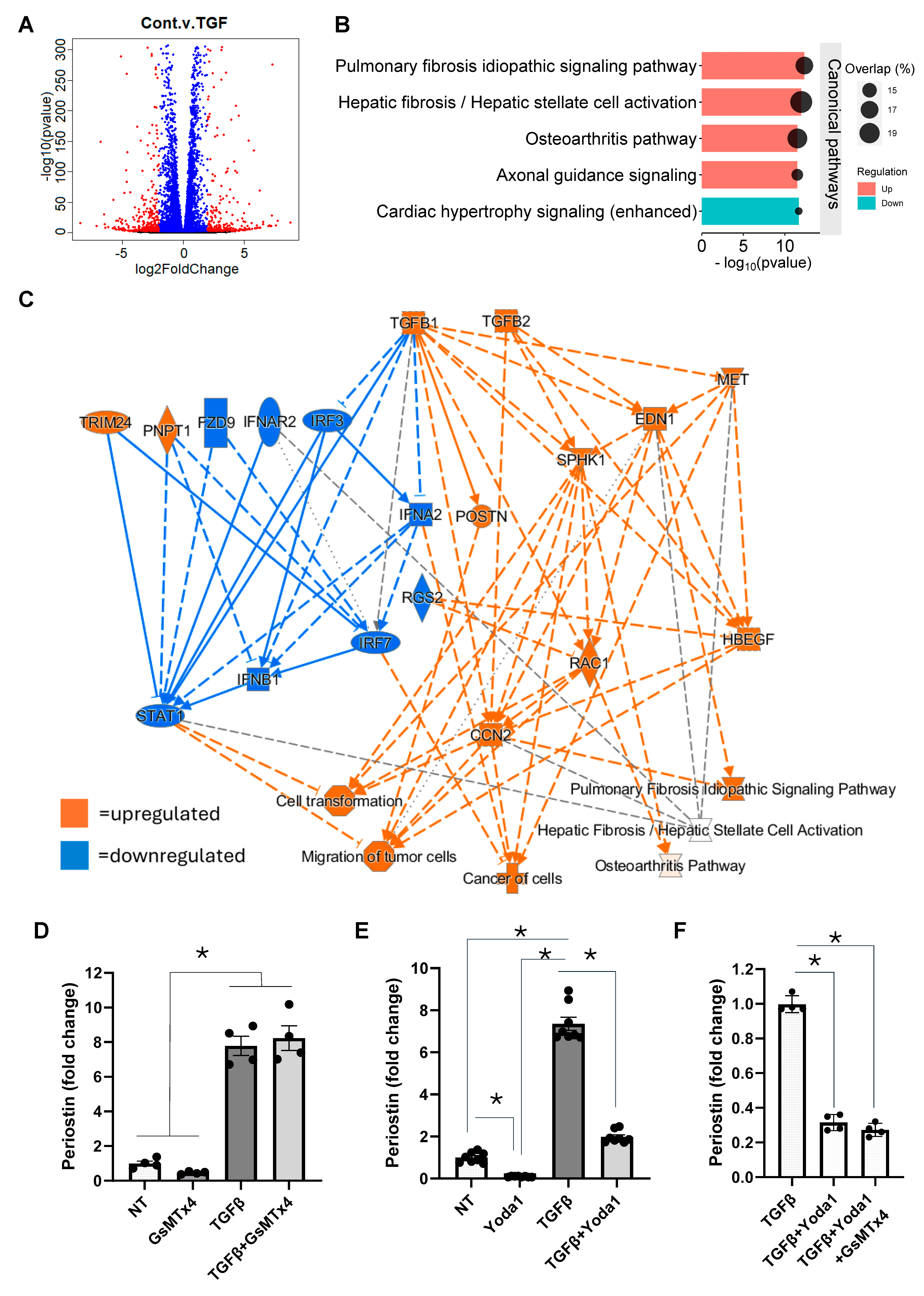
4.2. Yoda1 Inhibits Fibroblast Activation Through a Piezo1-Independent Mechanism
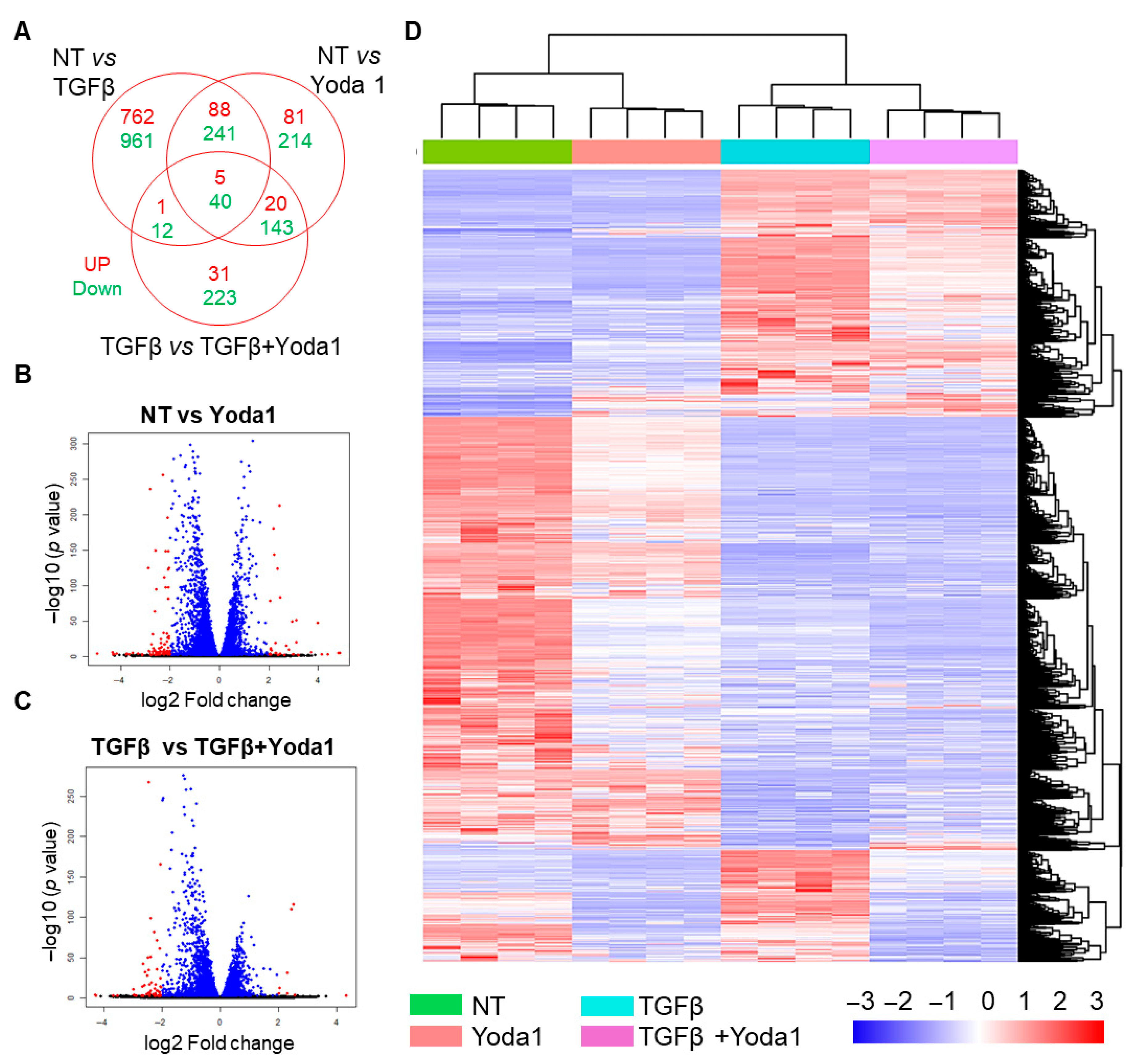
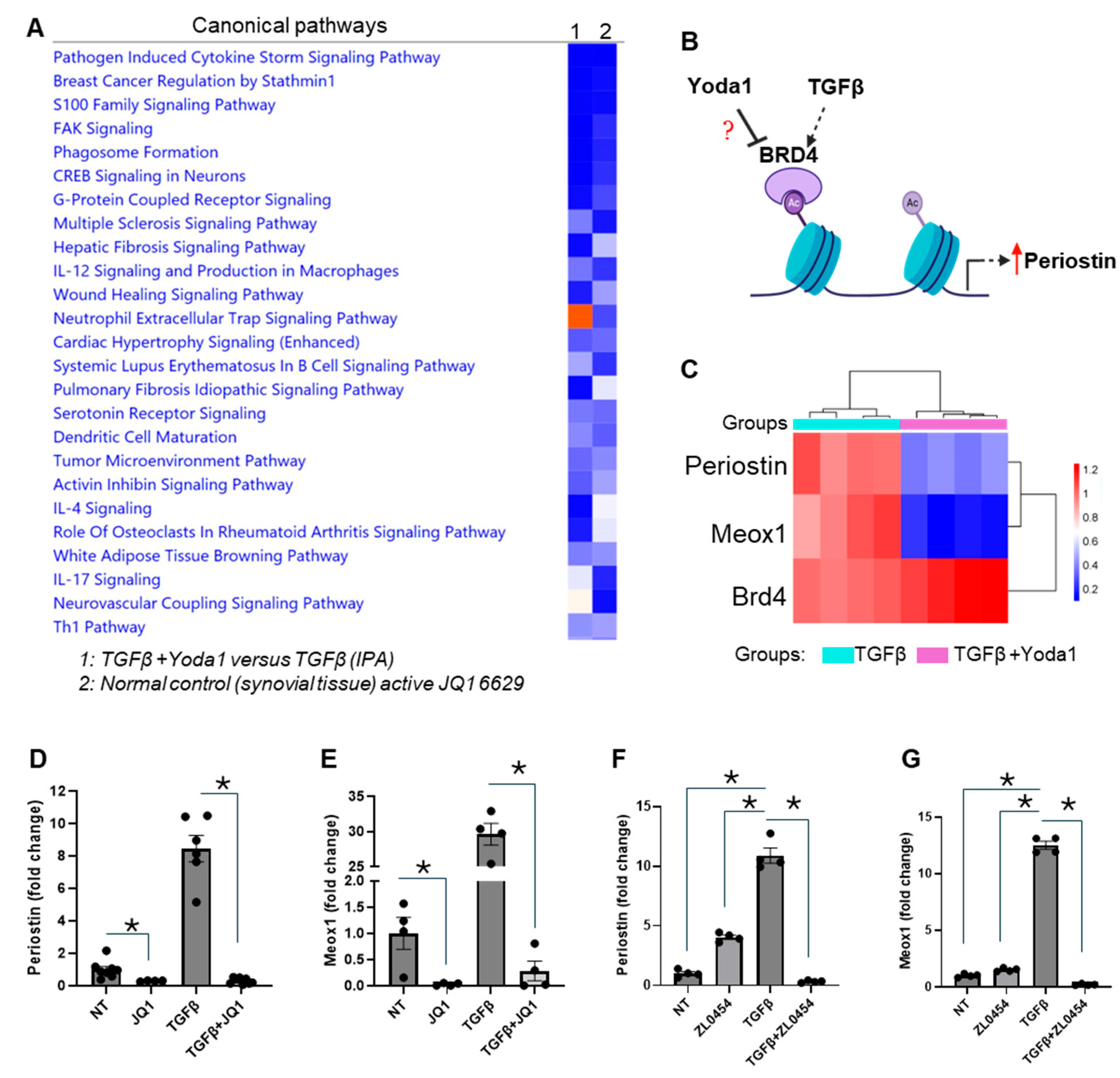
4.3. BRD4 Identified as a Potential Mediator of Yoda1-Induced Suppression of TGFβ-Induced Fibroblast Activation
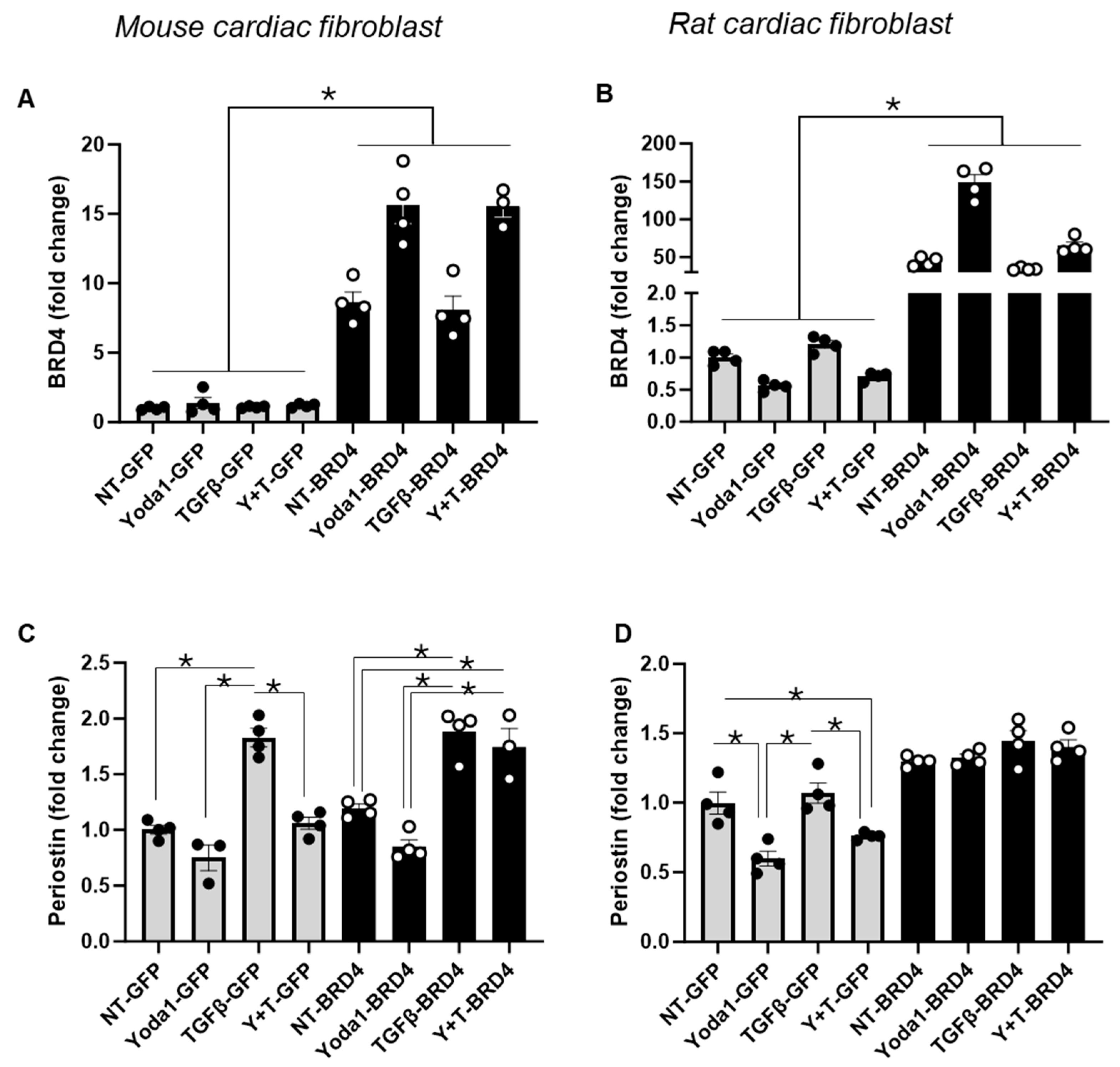
4.4. Overexpression of BRD4 Mitigates the Inhibitory Effect of Yoda1 on TGFβ-Induced Activation in Cardiac Fibroblasts
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J.; Tryggvason, K.; et al. Cardiomyopathy associated with microcirculation dysfunction in laminin alpha4 chain-deficient mice. J. Biol. Chem. 2006, 281, 213–220. [Google Scholar] [CrossRef]
- Michel, J.B. Anoikis in the cardiovascular system: Known and unknown extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154. [Google Scholar] [CrossRef]
- Rubart, M.; Tao, W.; Lu, X.L.; Conway, S.J.; Reuter, S.P.; Lin, S.F.; Soonpaa, M.H. Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart. Cardiovasc. Res. 2018, 114, 389–400. [Google Scholar] [CrossRef]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef]
- Dzeshka, M.S.; Lip, G.Y.; Snezhitskiy, V.; Shantsila, E. Cardiac Fibrosis in Patients With Atrial Fibrillation: Mechanisms and Clinical Implications. J. Am. Coll. Cardiol. 2015, 66, 943–959. [Google Scholar] [CrossRef]
- Martins, R.P.; Kaur, K.; Hwang, E.; Ramirez, R.J.; Willis, B.C.; Filgueiras-Rama, D.; Ennis, S.R.; Takemoto, Y.; Ponce-Balbuena, D.; Zarzoso, M.; et al. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 2014, 129, 1472–1482. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Sun, H.; Kerfant, B.G.; Crackower, M.A.; Penninger, J.M.; Backx, P.H. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell Cardiol. 2004, 37, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.H.; Wang, X.P.; Gao, C.Y.; Zhang, S.X.; Ma, X.H.; Liu, C. Blockade of KCa3.1 Attenuates Left Ventricular Remodeling after Experimental Myocardial Infarction. Cell Physiol. Biochem. 2015, 36, 1305–1315. [Google Scholar] [CrossRef]
- Teng, G.; Svystonyuk, D.; Mewhort, H.E.; Turnbull, J.D.; Belke, D.D.; Duff, H.J.; Fedak, P.W. Tetrandrine reverses human cardiac myofibroblast activation and myocardial fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1564–H1574. [Google Scholar] [CrossRef]
- Zhao, L.M.; Wang, L.P.; Wang, H.F.; Ma, X.Z.; Zhou, D.X.; Deng, X.L. The role of KCa3.1 channels in cardiac fibrosis induced by pressure overload in rats. Pflugers Arch. 2015, 467, 2275–2285. [Google Scholar] [CrossRef]
- Ramires, F.J.; Sun, Y.; Weber, K.T. Myocardial fibrosis associated with aldosterone or angiotensin II administration: Attenuation by calcium channel blockade. J. Mol. Cell Cardiol. 1998, 30, 475–483. [Google Scholar] [CrossRef]
- Roach, K.M.; Bradding, P. Ca2+ signalling in fibroblasts and the therapeutic potential of KCa3.1 channel blockers in fibrotic diseases. J. Pharmacol. 2020, 177, 1003–1024. [Google Scholar] [CrossRef]
- Anumanthan, G.; Wilson, P.J.; Tripathi, R.; Hesemann, N.P.; Mohan, R.R. Blockade of KCa3.1: A novel target to treat TGF-β1 induced conjunctival fibrosis. Exp. Eye Res. 2018, 167, 140–144. [Google Scholar] [CrossRef]
- Sforna, L.; Megaro, A.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. Structure, Gating and Basic Functions of the Ca2+-activated K Channel of Intermediate Conductance. Curr. Neuropharmacol. 2018, 16, 608–617. [Google Scholar] [CrossRef]
- Zhao, L.M.; Zhang, W.; Wang, L.P.; Li, G.R.; Deng, X.L. Advanced glycation end products promote proliferation of cardiac fibroblasts by upregulation of KCa3.1 channels. Pflugers Arch. 2012, 464, 613–621. [Google Scholar] [CrossRef]
- Zhao, L.M.; Su, X.L.; Wang, Y.; Li, G.R.; Deng, X.L. KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation endproducts in cultured rat vascular smooth muscle cells. Lab. Investig. 2013, 93, 159–167. [Google Scholar] [CrossRef] [PubMed]
- She, G.; Ren, Y.J.; Wang, Y.; Hou, M.C.; Wang, H.F.; Gou, W.; Lai, B.C.; Lei, T.; Du, X.J.; Deng, X.L. KCa3.1 Channels Promote Cardiac Fibrosis Through Mediating Inflammation and Differentiation of Monocytes Into Myofibroblasts in Angiotensin II–Treated Rats. J. Am. Heart Assoc. 2019, 8, e010418. [Google Scholar] [CrossRef]
- Stewart, L.; Turner, N.A. Channelling the Force to Reprogram the Matrix: Mechanosensitive Ion Channels in Cardiac Fibroblasts. Cells 2021, 10, 990. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Zhang, Z.; Tsujikawa, H.; Fusco, D.; Silverman, D.; Liang, B.; Yue, L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 2010, 106, 992–1003. [Google Scholar] [CrossRef]
- Trial, J.; Cieslik, K.A. Changes in cardiac resident fibroblast physiology and phenotype in aging. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H745–H755. [Google Scholar] [CrossRef] [PubMed]
- Ploeg, M.C.; Munts, C.; Prinzen, F.W.; Turner, N.A.; van Bilsen, M.; van Nieuwenhoven, F.A. Piezo1 Mechanosensitive Ion Channel Mediates Stretch-Induced Nppb Expression in Adult Rat Cardiac Fibroblasts. Cells 2021, 10, 1745. [Google Scholar] [CrossRef]
- Givan, S.A.; Bottoms, C.A.; Spollen, W.G. Computational analysis of RNA-seq. Methods Mol. Biol. 2012, 883, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, M.H.; Padilla, J.; Jenkins, N.T.; Thorne, P.K.; Martin, J.S.; Rector, R.S.; Akter, S.; Davis, J.W. Exercise-induced differential changes in gene expression among arterioles of skeletal muscles of obese rats. J. Appl. Physiol. (1985) 2015, 119, 583–603. [Google Scholar] [CrossRef]
- Laughlin, M.H.; Padilla, J.; Jenkins, N.T.; Thorne, P.K.; Martin, J.S.; Rector, R.S.; Akter, S.; Davis, J.W. Exercise training causes differential changes in gene expression in diaphragm arteries and 2A arterioles of obese rats. J. Appl. Physiol. (1985) 2015, 119, 604–616. [Google Scholar] [CrossRef]
- Padilla, J.; Simmons, G.H.; Davis, W.J.; Wyte, J.J.; Zderic, T.W.; Hamilton, M.T.; Bowles, D.K.; Laughlin, M.H. Impact of exercise training on endothelial transcriptional profiles in healthy swine: A genome-wide microarray analysis. J. Appl. Physiol. 2011, 301, H555–H564. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Jenkins, N.T.; Thorne, P.K.; Martin, J.S.; Rector, R.S.; Davis, J.W.; Laughlin, M.H. Transcriptome-wide RNA sequencing analysis of rat skeletal muscle feed arteries. II. Impact of exercise training in obesity. J. Appl. Physiol. (1985) 2014, 116, 1033–1047. [Google Scholar] [CrossRef]
- Jenkins, N.T.; Padilla, J.; Thorne, P.K.; Martin, J.S.; Rector, R.S.; Davis, J.W.; Laughlin, M.H. Transcriptome-wide RNA sequencing analysis of rat skeletal muscle feed arteries. I. Impact of obesity. J. Appl. Physiol. (1985) 2014, 116, 1017–1032. [Google Scholar] [CrossRef]
- Shi-Wen, X.; Leask, A.; Abraham, D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor. Rev. 2008, 19, 133–144. [Google Scholar] [CrossRef]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Kleniewska, P.; Kolodziejczyk, M.; Skibska, B.; Goraca, A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch. Immunol. Ther. Exp. 2015, 63, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Argentino, G.; Barbieri, A.; Beri, R.; Bason, C.; Ruzzenente, A.; Olivieri, O.; Tinazzi, E.; Puccetti, A.; Vitali, C.; Del Papa, N.; et al. Profibrotic Effects of Endothelin-1 on Fibroblasts Are Mediated by Aldosterone in Vitro: Relevance to the Pathogenesis and Therapy of Systemic Sclerosis and Pulmonary Arterial Hypertension. Biomedicines 2022, 10, 2765. [Google Scholar] [CrossRef]
- Petrini, I. Biology of MET: A double life between normal tissue repair and tumor progression. Ann. Transl. Med. 2015, 3, 82. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair. 2012, 5, 15. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef]
- Koselski, M.; Olszewska, A.; Hordyjewska, A.; Malecka-Massalska, T.; Trebacz, K. Three types of ion channels in the cell membrane of mouse fibroblasts. Physiol. Res. 2017, 66, 63–73. [Google Scholar] [CrossRef]
- Xing, C.; Bao, L.; Li, W.; Fan, H. Progress on role of ion channels of cardiac fibroblasts in fibrosis. Front. Physiol. 2023, 14, 1138306. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Xia, L. Excitation-contraction coupling between human atrial myocytes with fibroblasts and stretch activated channel current: A simulation study. Comput. Math. Methods Med. 2013, 2013, 238676. [Google Scholar] [CrossRef]
- Grandi, E.; Sanguinetti, M.C.; Bartos, D.C.; Bers, D.M.; Chen-Izu, Y.; Chiamvimonvat, N.; Colecraft, H.M.; Delisle, B.P.; Heijman, J.; Navedo, M.F.; et al. Potassium channels in the heart: Structure, function and regulation. J. Physiol. 2017, 595, 2209–2228. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Oda, S.; Shimauchi, T.; Nishimura, A.; Mangmool, S.; Nishida, M. TRPC3 Channels in Cardiac Fibrosis. Front. Cardiovasc. Med. 2017, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, N.; Watanabe, K.; Morimoto, S.; Sato, Y.; Kiyonaka, S.; Hoshijima, M.; Ikeda, Y.; Nakaya, M.; Ide, T.; Mori, Y.; et al. TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem. Biophys. Res. Commun. 2011, 409, 108–113. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 2016, 6, 39383. [Google Scholar] [CrossRef]
- Wang, L.P.; Wang, Y.; Zhao, L.M.; Li, G.R.; Deng, X.L. Angiotensin II upregulates K(Ca)3.1 channels and stimulates cell proliferation in rat cardiac fibroblasts. Biochem. Pharmacol. 2013, 85, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.R.; Shamhart, P.E.; Naugle, J.E.; Meszaros, J.G. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension 2008, 51, 704–711. [Google Scholar] [CrossRef]
- Campbell, S.E.; Katwa, L.C. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell Cardiol. 1997, 29, 1947–1958. [Google Scholar] [CrossRef]
- Duchemin, A.L.; Vignes, H.; Vermot, J. Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. eLife 2019, 8, e44706. [Google Scholar] [CrossRef]
- Braidotti, N.; Chen, S.N.; Long, C.S.; Cojoc, D.; Sbaizero, O. Piezo1 Channel as a Potential Target for Hindering Cardiac Fibrotic Remodeling. Int. J. Mol. Sci. 2022, 23, 8065. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, Y.; Cheng, X.; Hu, B.; Jiang, D.; Wu, L.; Peng, S.; Hu, J. Mechanotransductive receptor. Front. Mol. Biosci. 2023, 10, 1270979. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, H.; Zhang, M.; Liu, W.; Deng, T.; Zhao, Q.; Li, Y.; Lei, J.; Li, X.; Xiao, B. Structure and mechanogating of the mammalian tactile channel PIEZO2. Nature 2019, 573, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Braidotti, N.; Demontis, G.; Conti, M.; Andolfi, L.; Ciubotaru, C.D.; Sbaizero, O.; Cojoc, D. The local mechanosensitive response of primary cardiac fibroblasts is influenced by the microenvironment mechanics. Sci. Rep. 2024, 14, 10365. [Google Scholar] [CrossRef]
- Zhao, X.; Kong, Y.; Liang, B.; Xu, J.; Lin, Y.; Zhou, N.; Li, J.; Jiang, B.; Cheng, J.; Li, C.; et al. Mechanosensitive Piezo1 channels mediate renal fibrosis. JCI Insight 2022, 7, 152330. [Google Scholar] [CrossRef]
- Wang, L.; Wu, X.; Huang, P.; Lv, Z.; Qi, Y.; Wei, X.; Yang, P.; Zhang, F. JQ1, a small molecule inhibitor of BRD4, suppresses cell growth and invasion in oral squamous cell carcinoma. Oncol. Rep. 2016, 36, 1989–1996. [Google Scholar] [CrossRef]
- Wernersson, S.; Bobby, R.; Flavell, L.; Milbradt, A.G.; Holdgate, G.A.; Embrey, K.J.; Akke, M. Bromodomain Interactions with Acetylated Histone 4 Peptides in the BRD4 Tandem Domain: Effects on Domain Dynamics and Internal Flexibility. Biochemistry 2022, 61, 2303–2318. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, Z.C.; Wu, Y.Y.; Pi, Y.N.; Lou, S.H.; Liu, T.B.; Lou, G.; Yang, C. Bromodomain and extraterminal (BET) proteins: Biological functions, diseases, and targeted therapy. Signal Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef]
- Stratton, M.S.; Bagchi, R.A.; Felisbino, M.B.; Hirsch, R.A.; Smith, H.E.; Riching, A.S.; Enyart, B.Y.; Koch, K.A.; Cavasin, M.A.; Alexanian, M.; et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019, 125, 662–677. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Concentration | Company/Catalog Number |
|---|---|---|
| TGFβ | 10 ng/mL | Gibco™ (Waltham, MA, USA)/PHG9204 |
| Yoda1 | 10 µM | Sigma-Aldrich (St. Louis, MO, USA)/SML1558-5MG |
| TEA | 10 mM | Sigma-Aldrich (St. Louis, MO, USA)/8083520100 |
| 4-AP | 2 mM | Sigma-Aldrich (St. Louis, MO, USA)/A78403-25G |
| Ibtx | 100 nM | Tocris Bioscience™ (Minneapolis, MN, USA)/10-86100U |
| Apamin | 100 nM | Sigma-Aldrich (St. Louis, MO, USA)/A1289-1mg |
| TRAM34 | 100 nM | Sigma-Aldrich (St. Louis, MO, USA)/T6700-25mg |
| BaCl2 | 100 µM | Sigma-Aldrich (St. Louis, MO, USA)/B-0750 |
| GsMTx4 | 2.5 µM | Tocris Bioscience™ (Minneapolis, MN, USA)/49-121-00U |
| JQ1 | 10 µM | Sigma-Aldrich (St. Louis, MO, USA)/SML1524-5MG |
| ZL0454 | 10 µM | Medchemexpress LLC (Monmouth Junction, NJ, USA) /HY1121505MG |
| Gene Name | Species | Orientation | 5′-3′ Sequence |
|---|---|---|---|
| Periostin | Mouse | Forward | CCTGCCCTTATATGCTCTGCT |
| Mouse | Reverse | AAACATGGTCAATAGGCATCACT | |
| Meox1 | Mouse | Forward | GAAACCCCCACTCAGAAGATAGC |
| Mouse | Reverse | TCGTTGAAGATTCGCTCAGTC | |
| UBC | Mouse | Forward | CCCAGTGTTACCACCAAGAAG |
| Mouse | Reverse | CCCCATCACACCCAAGAACA | |
| BRD4 | Mouse | Forward | CTGCTCAGAGTGGTGCTCAA |
| Mouse | Reverse | TGCTGCCCTACCTGTTTCTT | |
| αSMA | Mouse | Forward | GCTTCGCTGGTGATGATGCTC |
| Mouse | Reverse | AGTTGGTGATGATGCCGTGTTC | |
| Periostin | Rat | Forward | TGCAAAAAGACACACCTGCAA |
| Rat | Reverse | CCGAAGTCAATGGGGCTCTT | |
| Meox1 | Rat | Forward | AGTTCGCCCACCATAACTACC |
| Rat | Reverse | TTGGAAAGGGCGTTCGTTCA | |
| UBC | Rat | Forward | ACACCAAGAAGGTCAAACAGGA |
| Rat | Reverse | GACACCTCCCCATCAAACCC |
| Gene | Channel Type | Channel Name | |
|---|---|---|---|
| 1 | Piezo1 | Cation | Piezo-type mechanosensitive ion channel component 1 |
| 2 | Vdac1 | Anion | Voltage-dependent anion channel 1 |
| 3 | Pkd2 | Cation | Polycystin 2, transient receptor potential cation channel |
| 4 | Trpm7 | Cation | Transient receptor potential cation channel, subfamily M, member 7 |
| 5 | Vdac2 | Anion | Voltage-dependent anion channel 2 |
| 6 | Vdac3 | Anion | Voltage-dependent anion channel 3 |
| 7 | Clns1a | Chloride | Chloride channel, nucleotide-sensitive, 1A |
| 8 | Cacna1c | Calcium | Calcium channel, voltage-dependent, L type, alpha 1C subunit |
| 9 | Clcn5 | Chloride | Chloride channel, voltage-sensitive 5 |
| 10 | Clcn4 | Chloride | Chloride channel, voltage-sensitive 4 |
| 11 | Cacna1g | Calcium | Calcium channel, voltage-dependent, T type, alpha 1G subunit |
| 12 | Cacng7 | Calcium | Calcium channel, voltage-dependent, gamma subunit 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, P.; Stiens, S.M.; Bowles, H.J.; Bui, H.; Bowles, D.K. Yoda1 Inhibits TGFβ-Induced Cardiac Fibroblast Activation via a BRD4-Dependent Pathway. Cells 2025, 14, 1028. https://doi.org/10.3390/cells14131028
Alam P, Stiens SM, Bowles HJ, Bui H, Bowles DK. Yoda1 Inhibits TGFβ-Induced Cardiac Fibroblast Activation via a BRD4-Dependent Pathway. Cells. 2025; 14(13):1028. https://doi.org/10.3390/cells14131028
Chicago/Turabian StyleAlam, Perwez, Sara M. Stiens, Hunter J. Bowles, Hieu Bui, and Douglas K. Bowles. 2025. "Yoda1 Inhibits TGFβ-Induced Cardiac Fibroblast Activation via a BRD4-Dependent Pathway" Cells 14, no. 13: 1028. https://doi.org/10.3390/cells14131028
APA StyleAlam, P., Stiens, S. M., Bowles, H. J., Bui, H., & Bowles, D. K. (2025). Yoda1 Inhibits TGFβ-Induced Cardiac Fibroblast Activation via a BRD4-Dependent Pathway. Cells, 14(13), 1028. https://doi.org/10.3390/cells14131028