
Transcriptomic Profiling of iPS Cell-Derived Hepatocyte-like Cells Reveals Their Close Similarity to Primary Liver Hepatocytes

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of TAV.iPSC and TAV.HLC Derivatives
2.2. RNAseq Sample Preparation
2.3. Quality Control, Alignment of Genomic DNA, and Differential Gene Analysis
2.4. Principal Component Analysis (PCA)
2.5. Pathway Enrichment Analysis
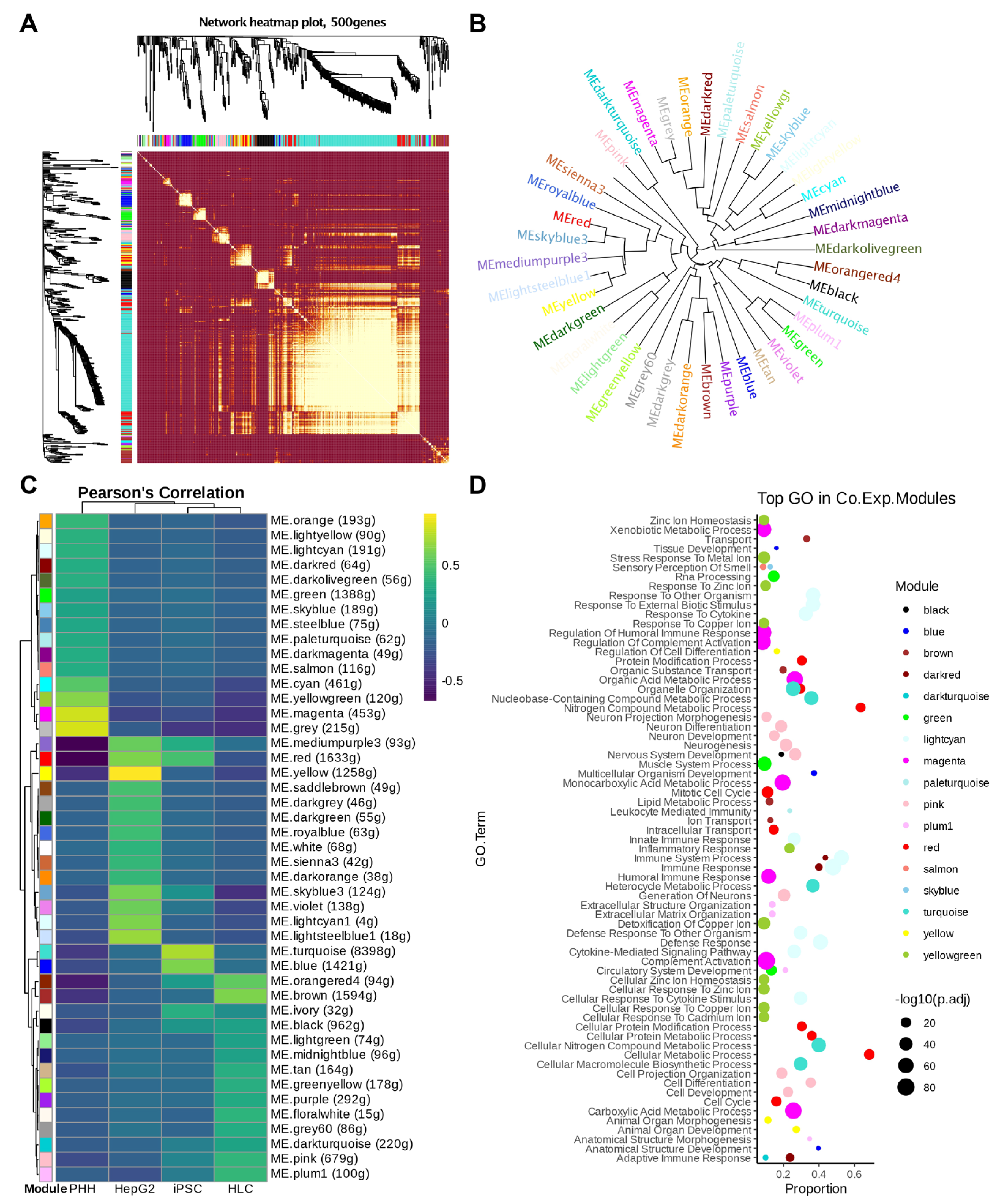
2.6. Weighted Correlation Network Analysis (WGCNA)
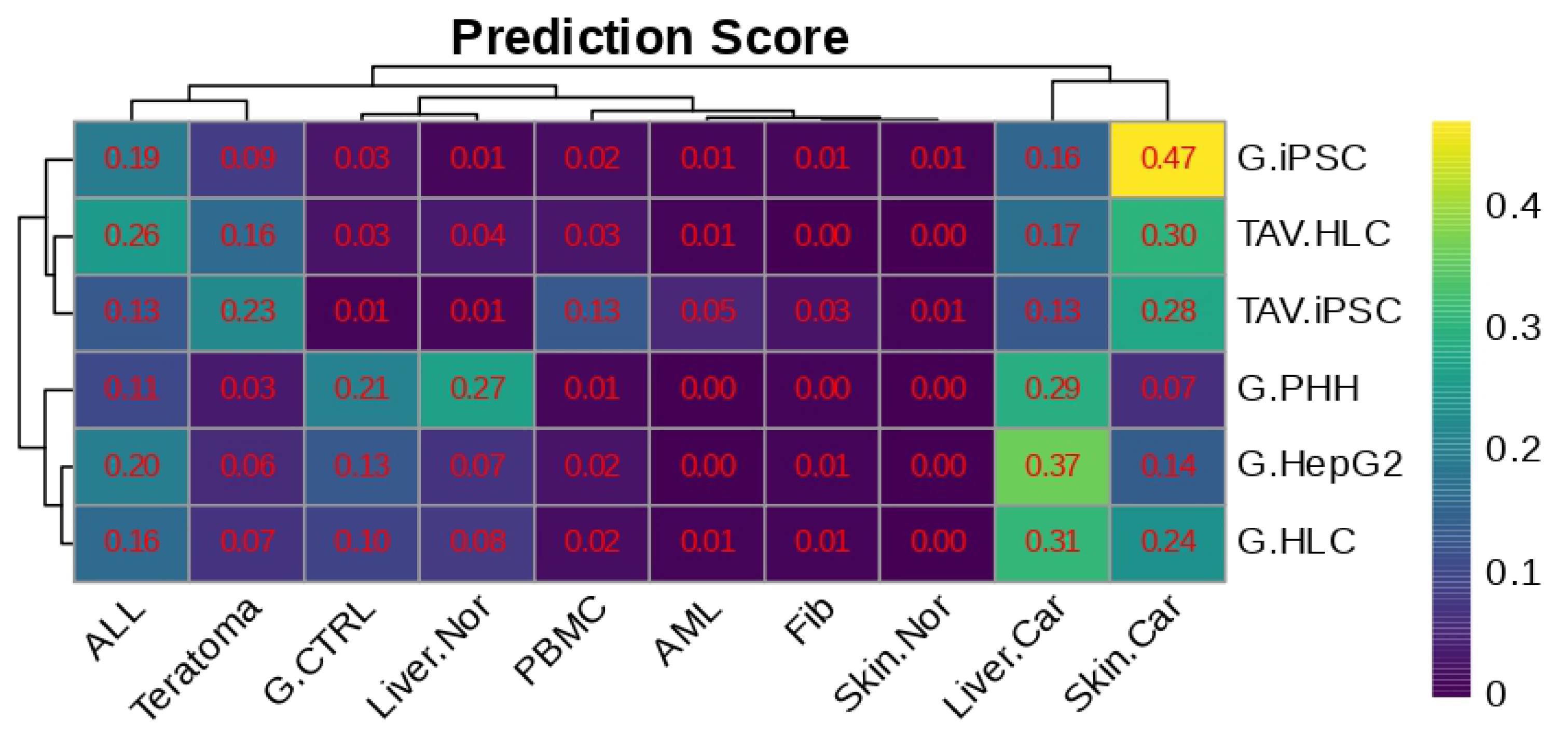
2.7. Phenotype Classification and Prediction
3. Results
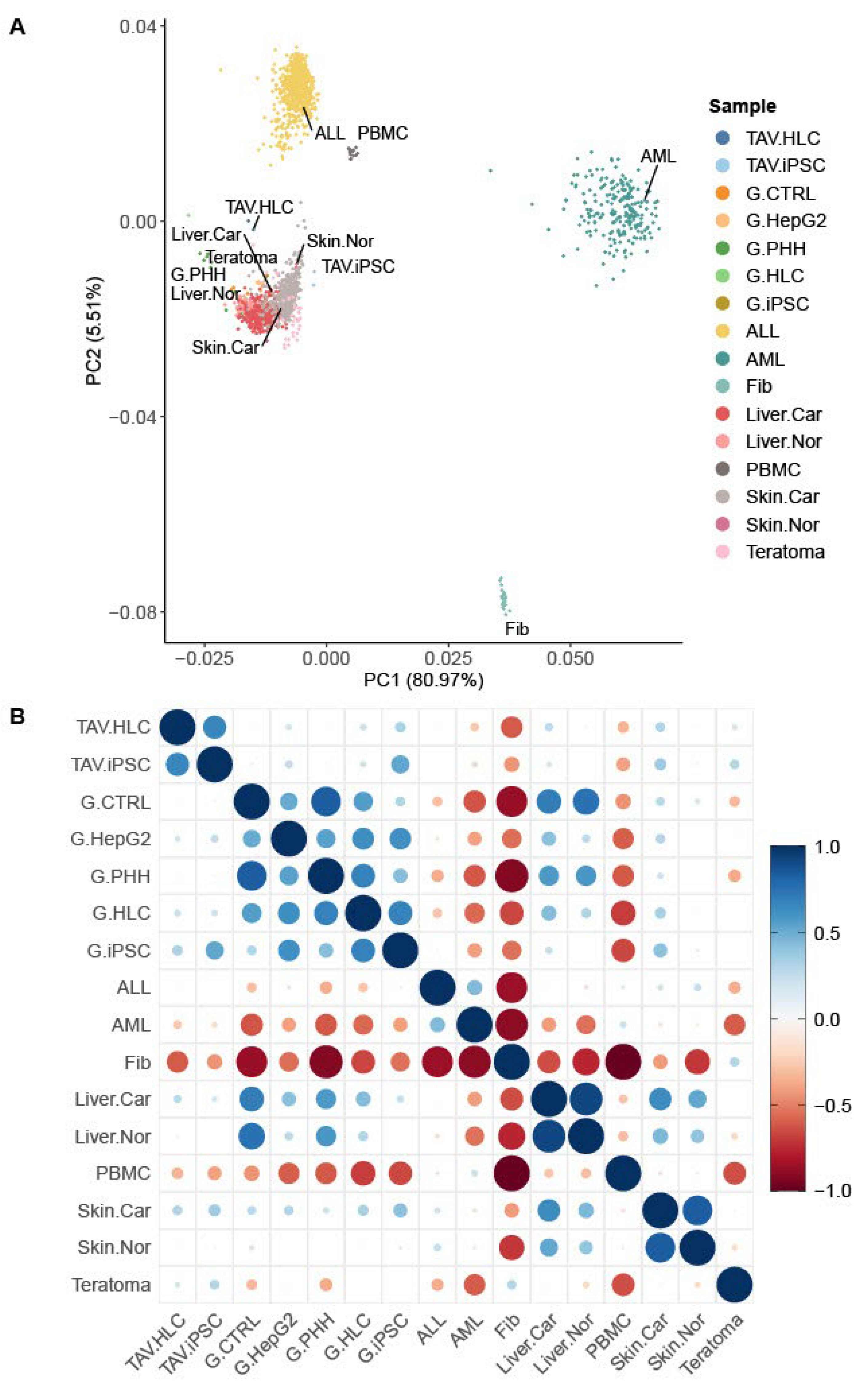
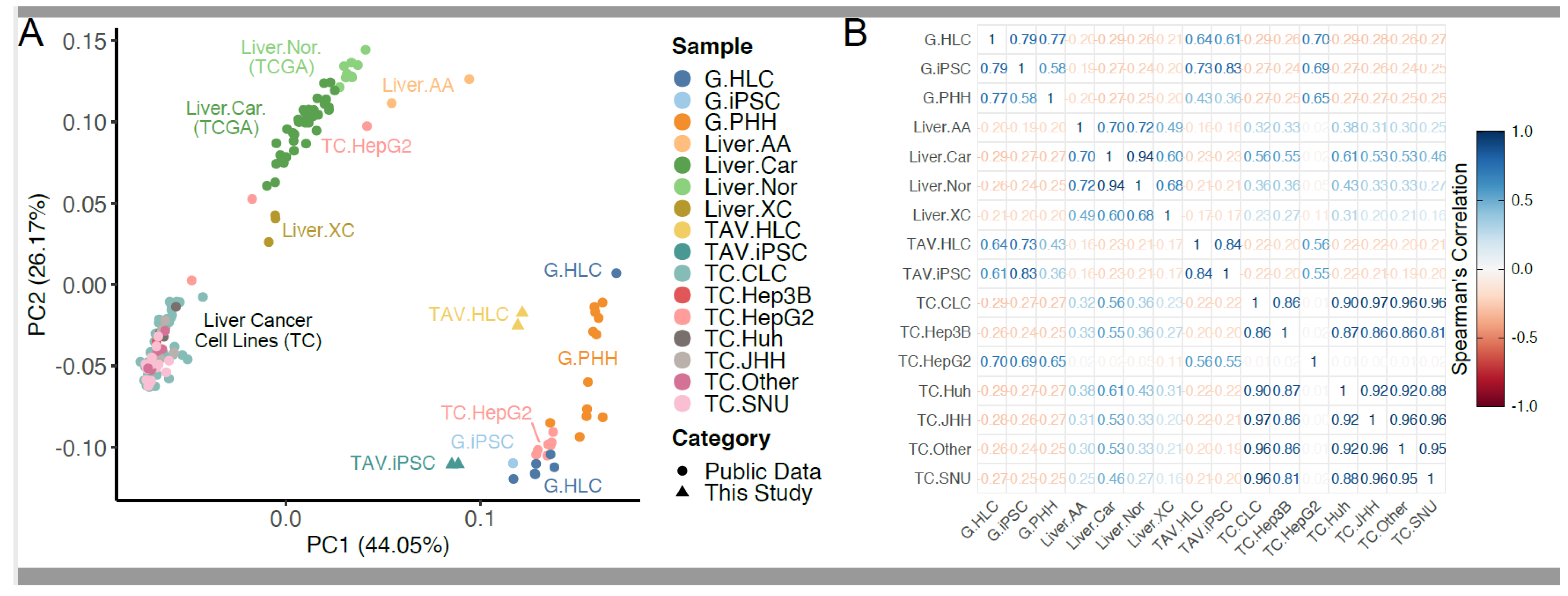
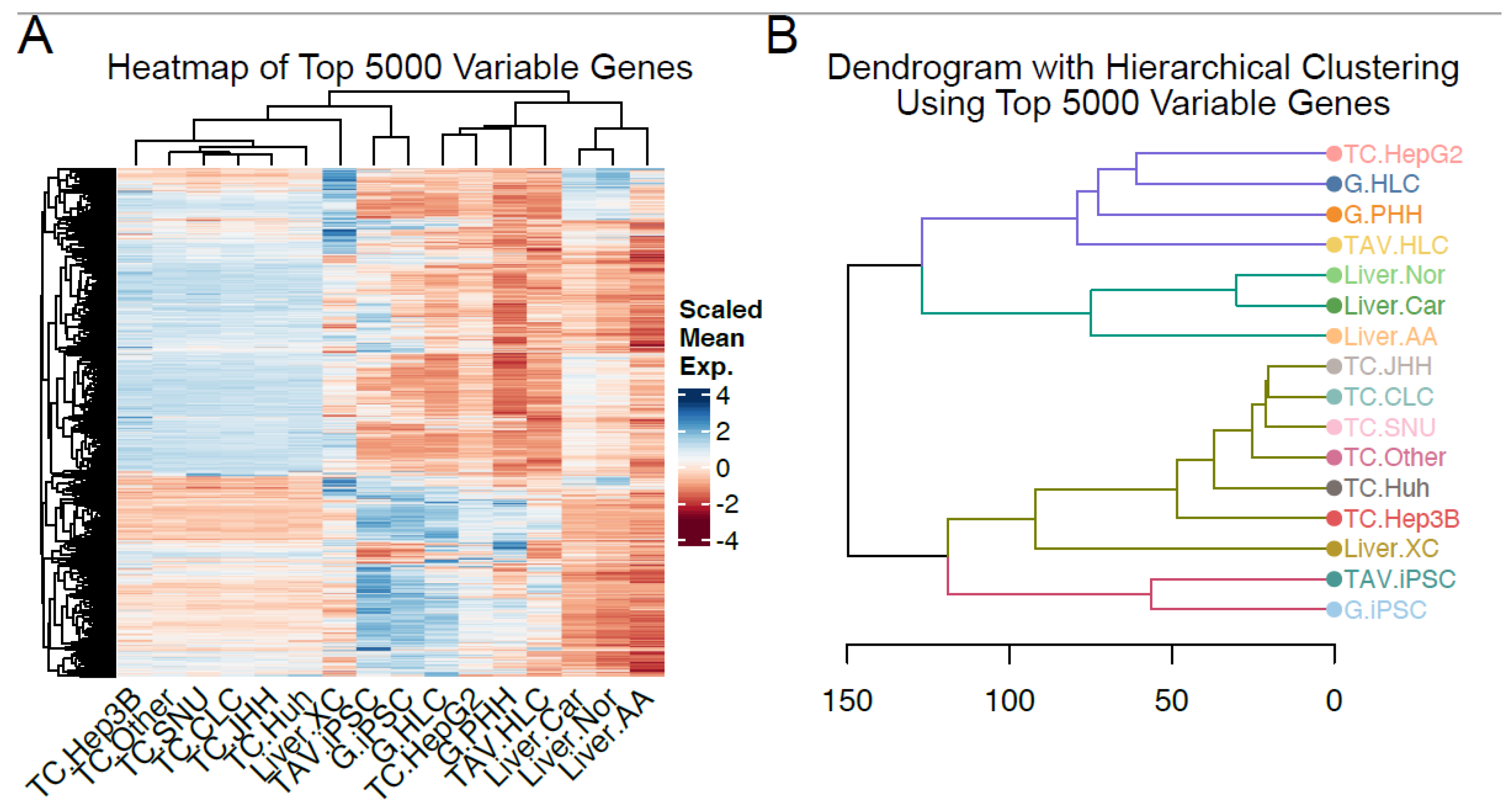
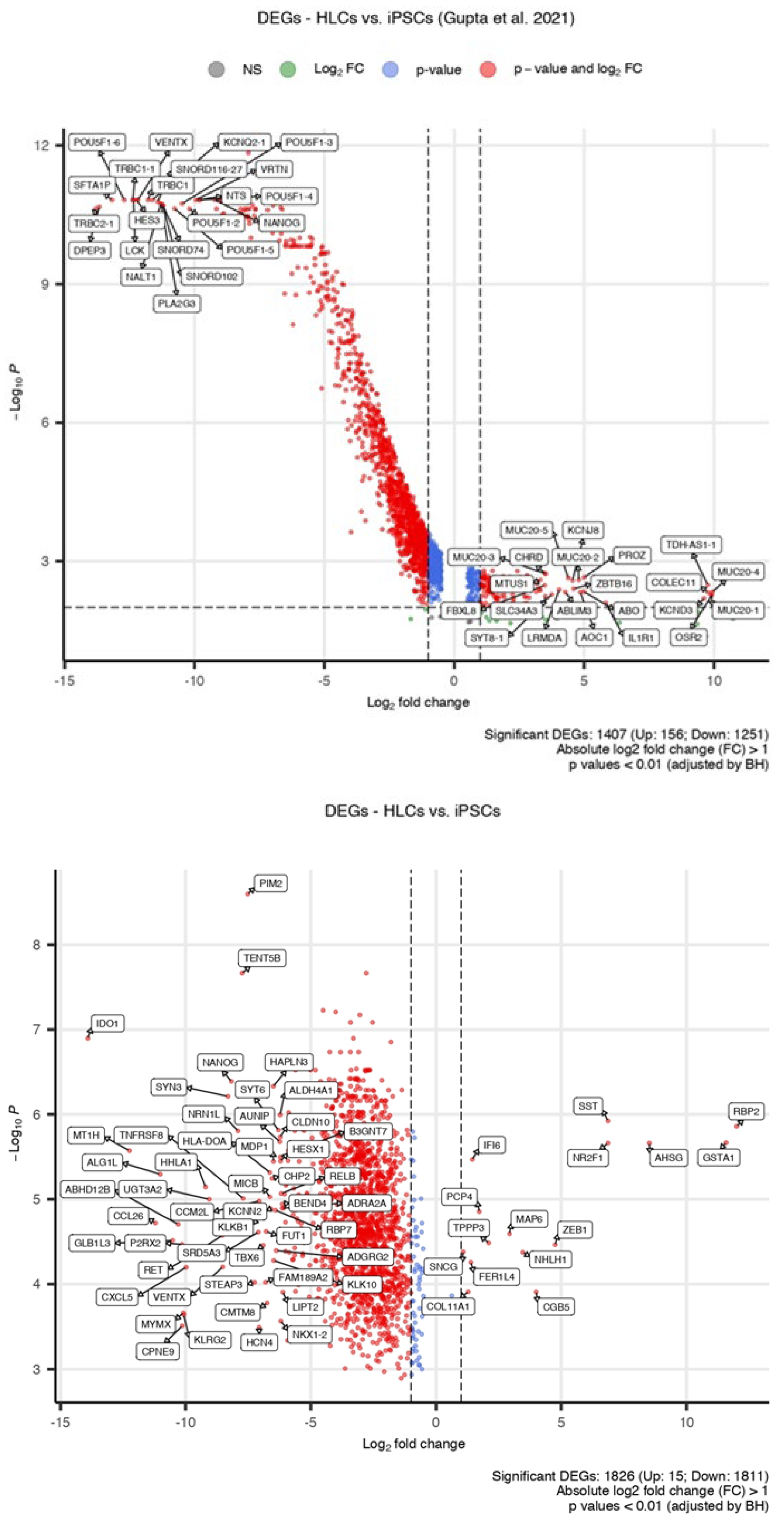
3.1. RNA-Seq Analysis Reveals Transcriptomic Similarity Between iPSC-Derived HLCs and PHHs
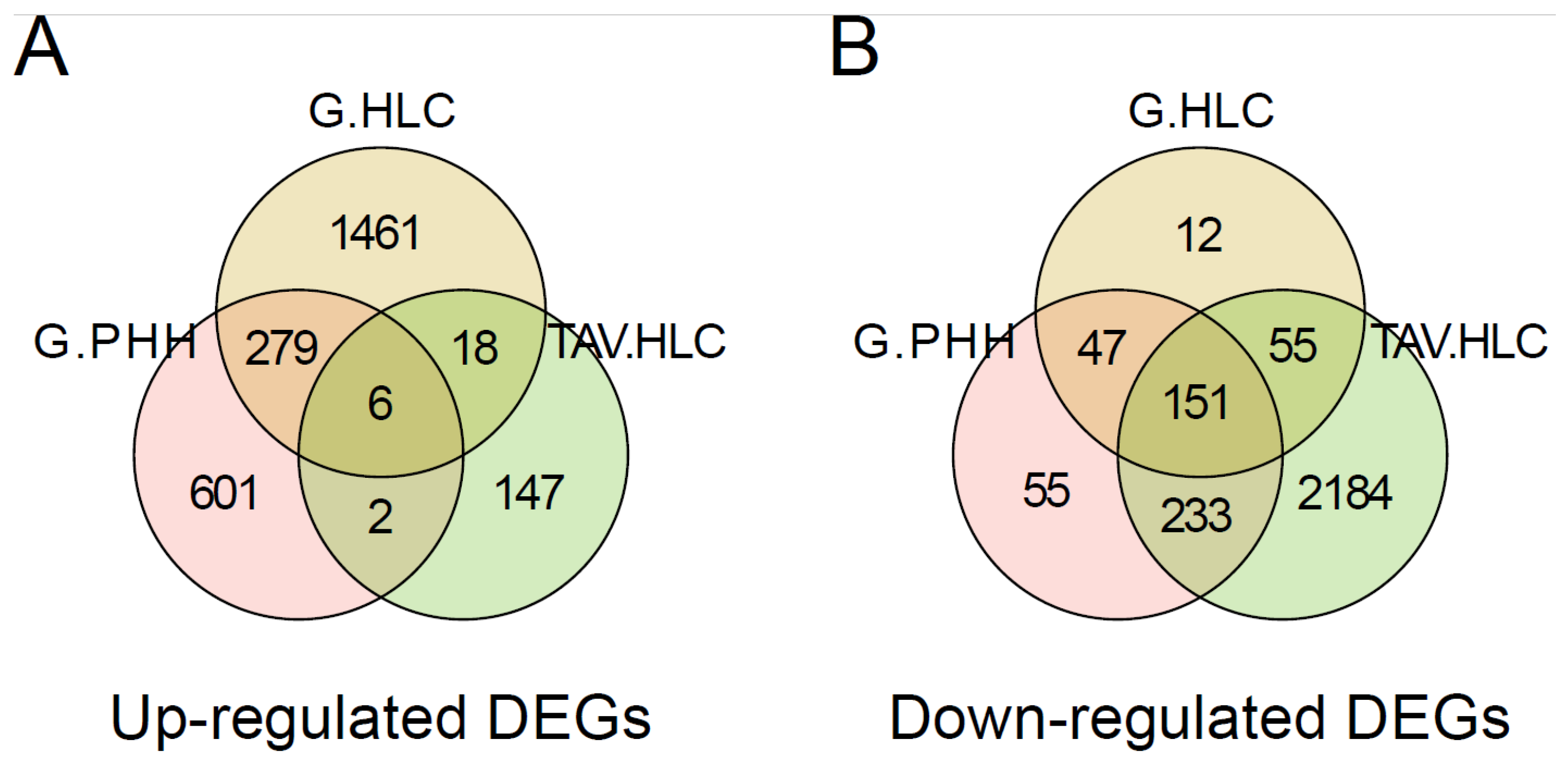
3.2. Pathway Analysis Shows Downregulated Genes Characteristic of Immune-Associated Signalling Pathways in iPSCs Compared to Those in HLCs
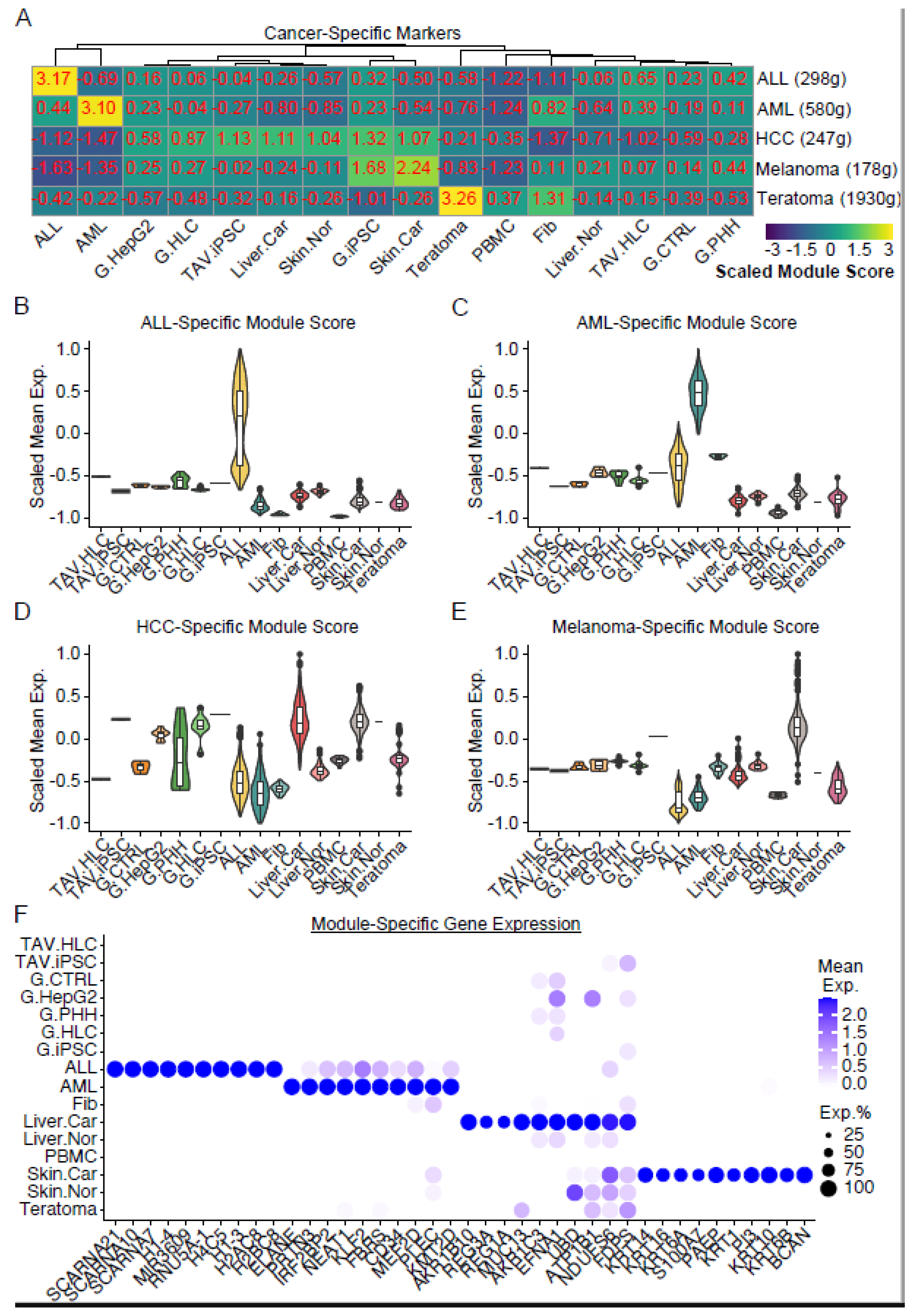
3.3. Analysis of Cancer Signatures in iPSCs or HLCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Huebert, R.C.; Rakela, J. Cellular therapy for liver disease. Mayo Clin. Proc. 2014, 89, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Georgomanoli, M.; Papapetrou, E.P. Modeling blood diseases with human induced pluripotent stem cells. Dis. Model. Mech. 2019, 12, 039321. [Google Scholar] [CrossRef]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar] [CrossRef]
- Chen, F.K.; McLenachan, S.; Edel, M.; Da Cruz, L.; Coffey, P.J.; Mackey, D.A. iPS Cells for Modelling and Treatment of Retinal Diseases. J. Clin. Med. 2014, 3, 1511–1541. [Google Scholar] [CrossRef]
- Burridge, P.W.; Keller, G.; Gold, J.D.; Wu, J.C. Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 2012, 10, 16–28. [Google Scholar] [CrossRef]
- Szkolnicka, D.; Farnworth, S.L.; Lucendo-Villarin, B.; Storck, C.; Zhou, W.; Iredale, J.P.; Flint, O.; Hay, D.C. Accurate prediction of drug-induced liver injury using stem cell-derived populations. Stem Cells Transl. Med. 2014, 3, 141–148. [Google Scholar] [CrossRef]
- Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Tucker, C.; Ferreira-Gonzalez, S.; Homer, N.; Wang, Y.; Starkey Lewis, P.J.; MToledo, E.; Mellado-Gomez, E.; Simpson, J.; et al. Dimethyl fumarate reduces hepatocyte senescence following paracetamol exposure. iScience 2021, 24, 102552. [Google Scholar] [CrossRef] [PubMed]
- Lucendo-Villarin, B.; Meseguer-Ripolles, J.; Drew, J.; Fischer, L.; Ma, E.; Flint, O.; Simpson, K.J.; Machesky, L.M.; Mountford, J.C.; Hay, D.C. Development of a cost-effective automated platform to produce human liver spheroids for basic and applied research. Biofabrication 2020, 13, 015009. [Google Scholar] [CrossRef] [PubMed]
- Esteve, J.; Blouin, J.M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Generation of induced pluripotent stem cells-derived hepatocyte-like cells for ex vivo gene therapy of primary hyperoxaluria type 1. Stem Cell Res. 2019, 38, 101467. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Tonnu, N.; Menon, T.; Lewis, B.M.; Green, K.T.; Wampler, D.; Monahan, P.E.; Verma, I.M. Autologous and Heterologous Cell Therapy for Hemophilia B toward Functional Restoration of Factor IX. Cell Rep. 2018, 23, 1565–1580. [Google Scholar] [CrossRef]
- Cameron, K.; Tan, R.; Schmidt-Heck, W.; Campos, G.; Lyall, M.J.; Wang, Y.; Lucendo-Villarin, B.; Szkolnicka, D.; Bates, N.; Kimber, S.J.; et al. Recombinant Laminins Drive the Differentiation and Self-Organization of hESC-Derived Hepatocytes. Stem Cell Rep. 2015, 5, 1250–1262. [Google Scholar] [CrossRef]
- Lucendo-Villarin, B.; Rashidi, H.; Alhaque, S.; Fischer, L.; Meseguer-Ripolles, J.; Wang, Y.; O’FArrelly, C.; Themis, M.; Hay, D.C. Serum Free Production of Three-dimensional Human Hepatospheres from Pluripotent Stem Cells. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Wang, Y.; Alhaque, S.; Cameron, K.; Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Rashidi, H.; Hay, D.C. Defined and Scalable Generation of Hepatocyte-like Cells from Human Pluripotent Stem Cells. J. Vis. Exp. 2017, 55355. [Google Scholar] [CrossRef]
- Rashidi, H.; Luu, N.T.; Alwahsh, S.M.; Ginai, M.; Alhaque, S.; Dong, H.; Tomaz, R.A.; Vernay, B.; Vigneswara, V.; Hallett, J.M.; et al. 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch. Toxicol. 2018, 92, 3117–3129. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Schrooders, Y.; Hauser, D.; van Herwijnen, M.; Albrecht, W.; Ter Braak, B.; Brecklinghaus, T.; Castell, J.V.; Elenschneider, L.; Escher, S.; et al. Comparing in vitro human liver models to in vivo human liver using RNA-Seq. Arch. Toxicol. 2021, 95, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Initiative ISC. Assessment of established techniques to determine developmental and malignant potential of human pluripotent stem cells. Nat. Commun. 2018, 9, 1925. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.G.; Wang, Z.; Kendziorski, C.; Thibeault, S.L. Tissue specific human fibroblast differential expression based on RNAsequencing analysis. BMC Genom. 2019, 20, 308. [Google Scholar] [CrossRef]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carre, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Nell, P.; Kattler, K.; Feuerborn, D.; Hellwig, B.; Rieck, A.; Salhab, A.; Lepikhov, K.; Gasparoni, G.; Thomitzek, A.; Belgasmi, K.; et al. Identification of an FXR-modulated liver-intestine hybrid state in iPSC-derived hepatocyte-like cells. J. Hepatol. 2022, 77, 1386–1398. [Google Scholar] [CrossRef]
- Jo, H.; Han, H.; Jung, I.; Ju, J.H.; Park, S.; Moon, S.; Geum, D.; Kim, H.; Park, H.; Kim, S.; et al. Development of genetic quality tests for good manufacturing practice-compliant induced pluripotent stem cells and their derivatives. Sci. Rep. 2020, 10, 3939. [Google Scholar] [CrossRef]
- Nghiem-Rao, T.H.; Pfeifer, C.; Asuncion, M.; Nord, J.; Schill, D.; Pulakanti, K.; Patel, S.B.; Cirillo, L.A.; Rao, S. Human induced pluripotent stem cell derived hepatocytes provide insights on parenteral nutrition associated cholestasis in the immature liver. Sci. Rep. 2021, 11, 12386. [Google Scholar] [CrossRef]
- Raggi, C.; M’Callum, M.; Pham, Q.T.; Gaub, P.; Selleri, S.; Baratang, N.V.; Mangahas, C.L.; Cagnone, G.; Reversade, B.; Joyal, J.; et al. Leveraging interacting signaling pathways to robustly improve the quality and yield of human pluripotent stem cell-derived hepatoblasts and hepatocytes. Stem Cell Rep. 2022, 17, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Chui, J.S.; Izuel-Idoype, T.; Qualizza, A.; de Almeida, R.P.; Piessens, L.; van der Veer, B.K.; Vanmarcke, G.; Malesa, A.; Athanasouli, P.; Boon, R.; et al. Osmolar Modulation Drives Reversible Cell Cycle Exit and Human Pluripotent Cell Differentiation via NF-κB and WNT Signaling. Adv. Sci. 2023, 11, 2307554. [Google Scholar] [CrossRef] [PubMed]
- Tascher, G.; Burban, A.; Camus, S.; Plumel, M.; Chanon, S.; Le Guevel, R.; Shevchenko, V.; Van Dorsselaer, A.; Lefai, E.; Guguen-Guillouzo, C.; et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells 2019, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Lucendo-Villarin, B.; Hay, D.C.; O’Farrelly, C. Human PSC-Derived Hepatocytes Express Low Levels of Viral Pathogen Recognition Receptors, but Are Capable of Mounting an Effective Innate Immune Response. Int. J. Mol. Sci. 2020, 21, 3831. [Google Scholar] [CrossRef]
- Horras, C.J.; Lamb, C.L.; Mitchell, K.A. Regulation of hepatocyte fate by interferon-gamma. Cytokine Growth Factor Rev. 2011, 22, 35–43. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Sample | Qty | Source | Data | Description |
|---|---|---|---|---|---|
| 1 | TAV.HLCs | 2 | In house | FPKM | Hepatocyte-like cells |
| 2 | TAV.iPSCs | 2 | In house | FPKM | Induced pluripotent stem cells |
| 3 | G.HepG2 | 7 | [22] | FastQ | Liver cancer cell line |
| 4 | G.PHHs | 12 | [22] | FastQ | Primary human hepatocytes |
| 5 | G.HLCs | 9 | [22] | FastQ | Hepatocyte-like cells |
| 6 | G.iPSCs | 2 | [22] | FastQ | Induced pluripotent stem cells |
| 7 | G.CTRL | 5 | [22] | FastQ | Primary human liver tissue—healthy |
| 8 | Liver-Nor | 50 | TCGA database | Count | Primary human liver tissue—normal |
| 9 | Liver-Car | 374 | TCGA database | Count | Primary human liver tissue—cancer |
| 10 | Skin-Nor | 1 | TCGA database | Count | Primary human skin tissue—normal |
| 11 | Skin-Car | 471 | TCGA database | Count | Primary human skin tissue—cancer |
| 12 | AML | 224 | TCGA database | Count | Acute myeloid leukaemia |
| 13 | ALL.P1-3 | 679 | TCGA/TARGET database | Count | Acute lymphocytic leukaemia phase 1–3 |
| 14 | Teratoma | 37 | [23] | FPKM | Teratoma |
| 15 | Fibroblast | 34 | [24] | FPKM | Tissue-specific fibroblasts |
| 16 | PBMCbulk | 13 | [25] | TPM | Peripheral blood mononuclear cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suleman, S.; Alhaque, S.; Guo, A.; Zhang, A.; Fawaz, S.; Perera, S.; Khalifa, M.S.; Rashidi, H.; Hay, D.C.; Themis, M. Transcriptomic Profiling of iPS Cell-Derived Hepatocyte-like Cells Reveals Their Close Similarity to Primary Liver Hepatocytes. Cells 2025, 14, 925. https://doi.org/10.3390/cells14120925
Suleman S, Alhaque S, Guo A, Zhang A, Fawaz S, Perera S, Khalifa MS, Rashidi H, Hay DC, Themis M. Transcriptomic Profiling of iPS Cell-Derived Hepatocyte-like Cells Reveals Their Close Similarity to Primary Liver Hepatocytes. Cells. 2025; 14(12):925. https://doi.org/10.3390/cells14120925
Chicago/Turabian StyleSuleman, Saqlain, Sharmin Alhaque, Andrew Guo, Aaron Zhang, Serena Fawaz, Stefany Perera, Mohammad S. Khalifa, Hassan Rashidi, David C. Hay, and Michael Themis. 2025. "Transcriptomic Profiling of iPS Cell-Derived Hepatocyte-like Cells Reveals Their Close Similarity to Primary Liver Hepatocytes" Cells 14, no. 12: 925. https://doi.org/10.3390/cells14120925
APA StyleSuleman, S., Alhaque, S., Guo, A., Zhang, A., Fawaz, S., Perera, S., Khalifa, M. S., Rashidi, H., Hay, D. C., & Themis, M. (2025). Transcriptomic Profiling of iPS Cell-Derived Hepatocyte-like Cells Reveals Their Close Similarity to Primary Liver Hepatocytes. Cells, 14(12), 925. https://doi.org/10.3390/cells14120925