

Downregulated ALDH2 Contributes to Tumor Progression and Targeted Therapy Resistance in Human Metastatic Melanoma Cells

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Bioinformatics Analysis
2.3. Cell Culture and AcAH Treatment
2.4. Quantitative RT-PCR
2.5. Western Blot
2.6. ALDH2 Knockout (KO)
2.7. ALDH2 Overexpression (OE)
2.8. ALDH2 Activity Assay
2.9. AcAH Assay
2.10. Cellular ROS Assay
2.11. Cellular Energy Metabolism Assay
2.12. Tumor Formation In Vivo
2.13. Interleukin (IL)-1β Secretion
2.14. Cell Viability Assay
2.15. Generation of Acquired Resistance
2.16. Statistical Analysis
3. Results
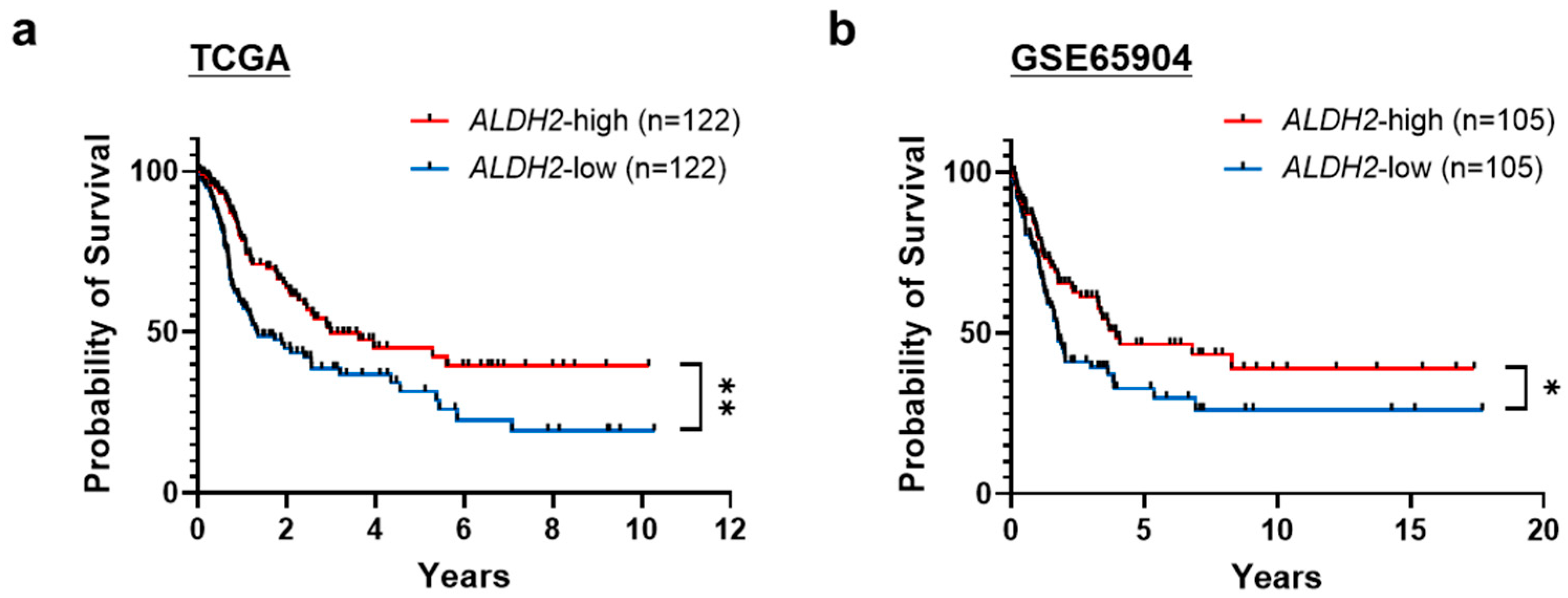
3.1. Low ALDH2 Expression Correlates with Worse Overall Survival in Metastatic Melanoma
3.2. Melanoma Cells Are Classified as ALDH2-Normal or ALDH2-Low, Compared to Normal Skin Cells
3.3. ALDH2-KO Impairs AcAH Detoxification and Alters Redox Balance and Cellular Energy Metabolism in ALDH2-Normal A375 Cells
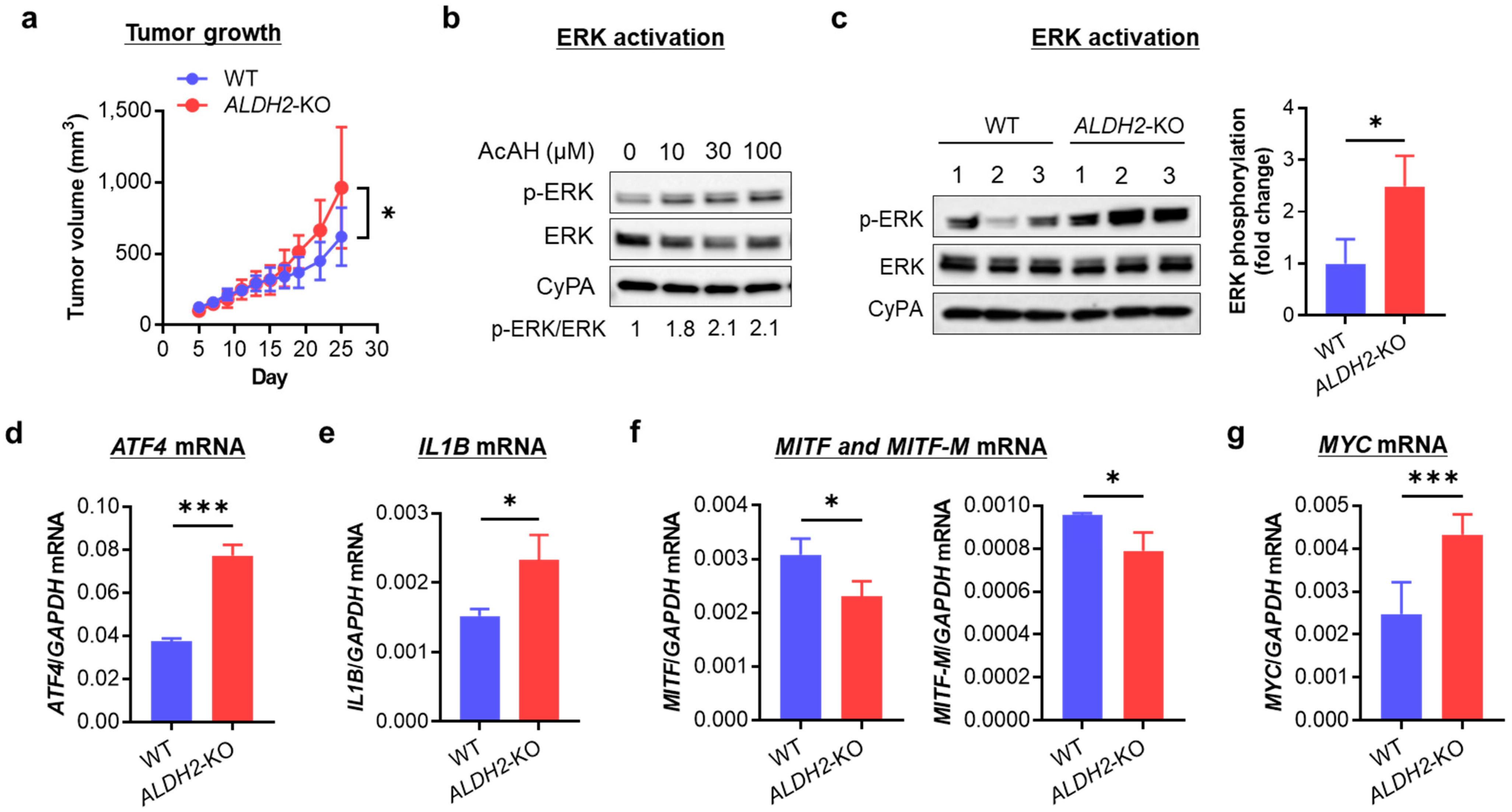
3.4. ALDH2 Downregulation Promotes Melanoma Tumor Growth by Enhancing AcAH-Mediated MAPK/ERK Activation, Inflammation, and Glycolytic Reprogramming
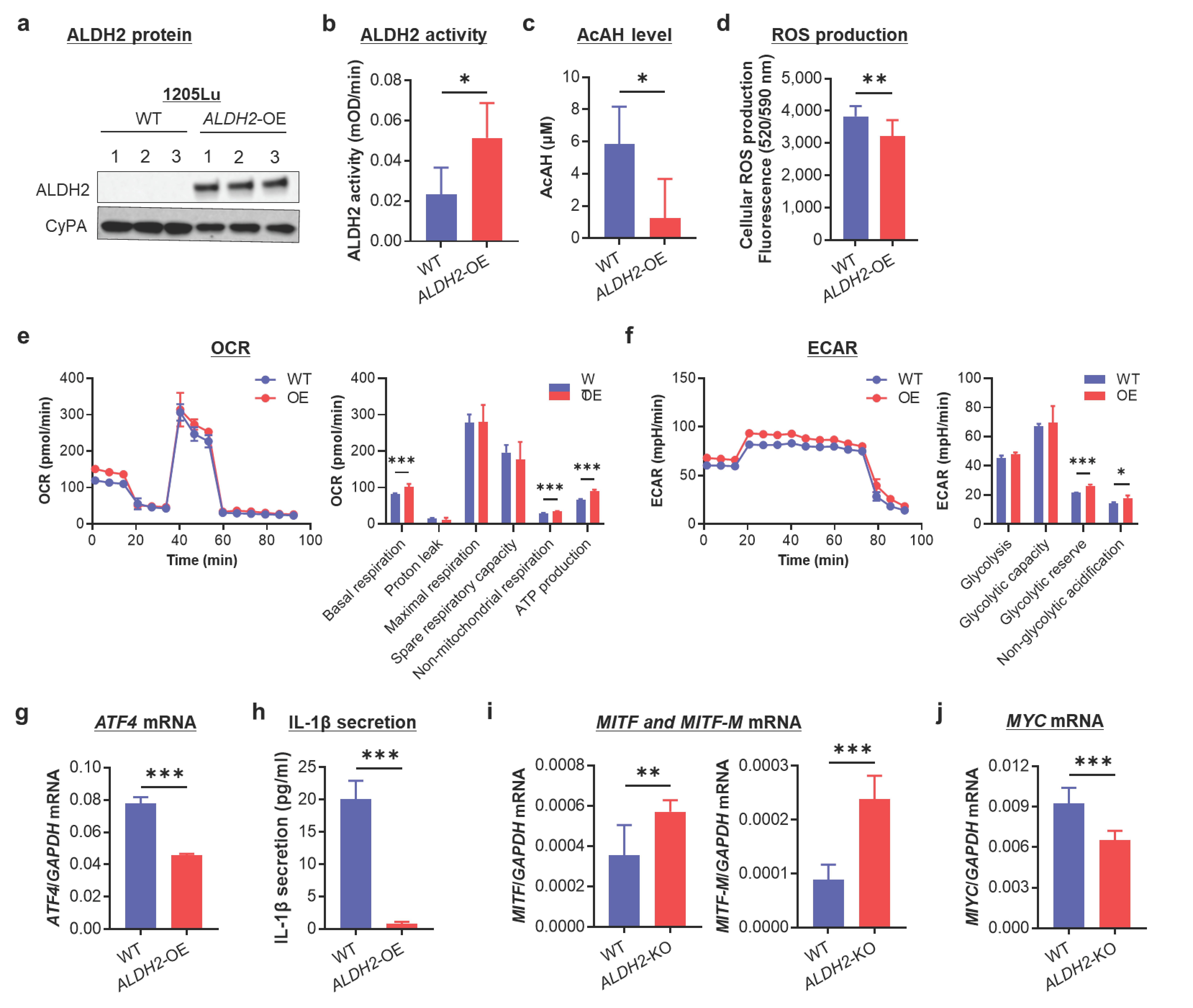
3.5. ALDH2-OE Alleviates AcAH, ROS, and IL-1β Production and Promotes Mitochondrial Respiration and a Metabolic Shift Toward Oxidative Phosphorylation in ALDH2-Low 1205Lu Cells
3.6. Melanoma Cells with Low ALDH2 Expression Harbor Intrinsic Resistance to MAPK/ERK Signaling Inhibition
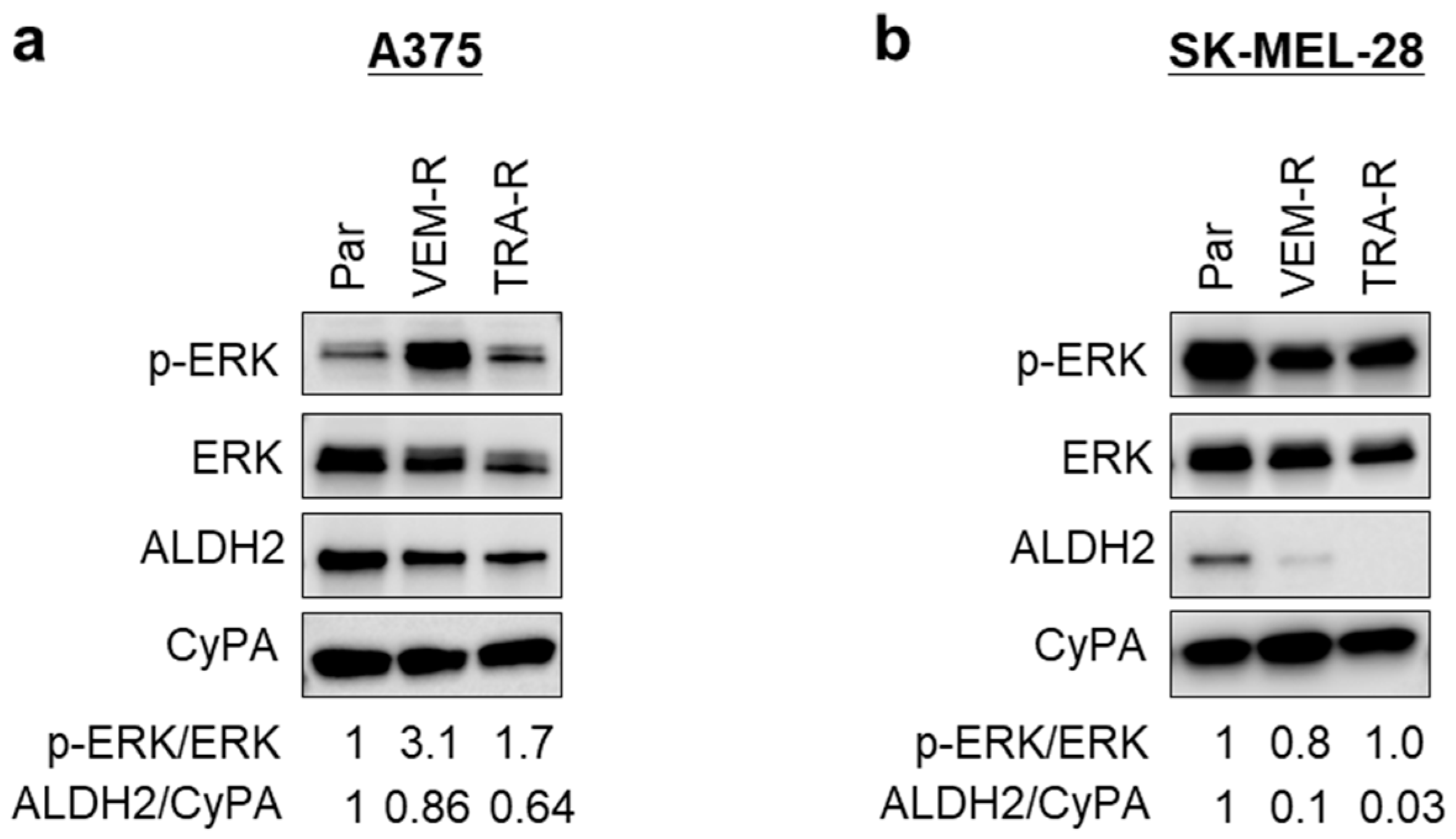
3.7. Acquired Resistance in Melanoma Cells Is Associated with Reduced ALDH2 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed]
- Lehner, T.; Gao, B.; Mackowiak, B. Alcohol metabolism in alcohol use disorder: A potential therapeutic target. Alcohol. Alcohol. 2024, 59, agad077. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hao, Y.; Piao, X.; Gu, X. Aldehyde Dehydrogenase 2 as a Therapeutic Target in Oxidative Stress-Related Diseases: Post-Translational Modifications Deserve More Attention. Int. J. Mol. Sci. 2022, 23, 2682. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Carresi, C.; Gliozzi, M.; Musolino, V.; Macri, R.; Scarano, F.; Coppoletta, A.; Cardamone, A.; Bosco, F.; et al. The Generation of Nitric Oxide from Aldehyde Dehydrogenase-2: The Role of Dietary Nitrates and Their Implication in Cardiovascular Disease Management. Int. J. Mol. Sci. 2022, 23, 15454. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef]
- Stewart, M.J.; Malek, K.; Crabb, D.W. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J. Investig. Med. 1996, 44, 42–46. [Google Scholar]
- Cheung, C.; Davies, N.G.; Hoog, J.O.; Hotchkiss, S.A.; Smith Pease, C.K. Species variations in cutaneous alcohol dehydrogenases and aldehyde dehydrogenases may impact on toxicological assessments of alcohols and aldehydes. Toxicology 2003, 184, 97–112. [Google Scholar] [CrossRef]
- Zhai, Z.; Yamauchi, T.; Shangraw, S.; Hou, V.; Matsumoto, A.; Fujita, M. Ethanol Metabolism and Melanoma. Cancers 2023, 15, 1258. [Google Scholar] [CrossRef]
- Matsumoto, A.; Ito, S.; Wakamatsu, K.; Ichiba, M.; Vasiliou, V.; Akao, C.; Song, B.J.; Fujita, M. Ethanol induces skin hyperpigmentation in mice with aldehyde dehydrogenase 2 deficiency. Chem. Biol. Interact. 2019, 302, 61–66. [Google Scholar] [CrossRef]
- Liu, L.; Chen, J. Advances in Relationship Between Alcohol Consumption and Skin Diseases. Clin. Cosmet. Investig. Dermatol. 2023, 16, 3785–3791. [Google Scholar] [CrossRef]
- Yamauchi, T.; Shangraw, S.; Zhai, Z.; Ravindran Menon, D.; Batta, N.; Dellavalle, R.P.; Fujita, M. Alcohol as a Non-UV Social-Environmental Risk Factor for Melanoma. Cancers 2022, 14, 5010. [Google Scholar] [CrossRef]
- Mahamat-Saleh, Y.; Al-Rahmoun, M.; Severi, G.; Ghiasvand, R.; Veierod, M.B.; Caini, S.; Palli, D.; Botteri, E.; Sacerdote, C.; Ricceri, F.; et al. Baseline and lifetime alcohol consumption and risk of skin cancer in the European Prospective Investigation into Cancer and Nutrition cohort (EPIC). Int. J. Cancer 2023, 152, 348–362. [Google Scholar] [CrossRef]
- Lei, H.; Liao, J.; Wang, X.; Huang, R.; Ying, C.; Yang, J. ALDH2 is a novel biomarker and exerts an inhibitory effect on melanoma. Sci. Rep. 2024, 14, 4183. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zhou, E.; Cheng, C. A B-Raf V600E gene signature for melanoma predicts prognosis and reveals sensitivity to targeted therapies. Cancer Med. 2022, 11, 1232–1243. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef] [PubMed]
- Imani, S.; Roozitalab, G.; Emadi, M.; Moradi, A.; Behzadi, P.; Jabbarzadeh Kaboli, P. The evolution of BRAF-targeted therapies in melanoma: Overcoming hurdles and unleashing novel strategies. Front. Oncol. 2024, 14, 1504142. [Google Scholar] [CrossRef]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma—A mini review. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef]
- Aroor, A.R.; Shukla, S.D. MAP kinase signaling in diverse effects of ethanol. Life Sci. 2004, 74, 2339–2364. [Google Scholar] [CrossRef]
- Lee, Y.J.; Aroor, A.R.; Shukla, S.D. Temporal activation of p42/44 mitogen-activated protein kinase and c-Jun N-terminal kinase by acetaldehyde in rat hepatocytes and its loss after chronic ethanol exposure. J. Pharmacol. Exp. Ther. 2002, 301, 908–914. [Google Scholar] [CrossRef]
- Oh, T.; Kwon, M.; Yu, J.S.; Jang, M.; Kim, G.H.; Kim, K.H.; Ko, S.K.; Ahn, J.S. Ent-Peniciherqueinone Suppresses Acetaldehyde-Induced Cytotoxicity and Oxidative Stress by Inducing ALDH and Suppressing MAPK Signaling. Pharmaceutics 2020, 12, 1229. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cirenajwis, H.; Ekedahl, H.; Lauss, M.; Harbst, K.; Carneiro, A.; Enoksson, J.; Rosengren, F.; Werner-Hartman, L.; Torngren, T.; Kvist, A.; et al. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 2015, 6, 12297–12309. [Google Scholar] [CrossRef] [PubMed]
- Jeong, I.; Yu, N.; Jang, I.; Jun, Y.; Kim, M.S.; Choi, J.; Lee, B.; Lee, S. GEMiCCL: Mining genotype and expression data of cancer cell lines with elaborate visualization. Database 2018, 2018, bay041. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Vaddi, P.K.; Samson, J.M.; Takegami, T.; Fujita, M. NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma. Pharmaceuticals 2020, 14, 23. [Google Scholar] [CrossRef]
- Rwere, F.; White, J.R.; Hell, R.C.R.; Yu, X.; Zeng, X.; McNeil, L.; Zhou, K.N.; Angst, M.S.; Chen, C.H.; Mochly-Rosen, D.; et al. Uncovering newly identified aldehyde dehydrogenase 2 genetic variants that lead to acetaldehyde accumulation after an alcohol challenge. J. Transl. Med. 2024, 22, 697. [Google Scholar] [CrossRef]
- Nannelli, G.; Terzuoli, E.; Giorgio, V.; Donnini, S.; Lupetti, P.; Giachetti, A.; Bernardi, P.; Ziche, M. ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid. Med. Cell Longev. 2018, 2018, 9765027. [Google Scholar] [CrossRef]
- Yan, T.; Zhao, Y. Acetaldehyde induces phosphorylation of dynamin-related protein 1 and mitochondrial dysfunction via elevating intracellular ROS and Ca2+ levels. Redox Biol. 2020, 28, 101381. [Google Scholar] [CrossRef]
- Lindros, K.O.; Stowell, A.; Pikkarainen, P.; Salaspuro, M. Elevated blood acetaldehyde in alcoholics with accelerated ethanol elimination. Pharmacol. Biochem. Behav. 1980, 13 (Suppl. 1), 119–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Yang, L.F.; Lai, C.L.; Yin, S.J. Acetaldehyde Enhances Alcohol Sensitivity and Protects against Alcoholism: Evidence from Alcohol Metabolism in Subjects with Variant ALDH2*2 Gene Allele. Biomolecules 2021, 11, 1183. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Radi, R.H.; Arbiser, J.L. Mitochondrial Metabolism in Melanoma. Cells 2021, 10, 3197. [Google Scholar] [CrossRef]
- Zhai, Z.; Samson, J.M.; Yamauchi, T.; Vaddi, P.K.; Matsumoto, Y.; Dinarello, C.A.; Ravindran Menon, D.; Fujita, M. Inflammasome Sensor NLRP1 Confers Acquired Drug Resistance to Temozolomide in Human Melanoma. Cancers 2020, 12, 2518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacol. Ther. 2011, 132, 86–95. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, B.; Zhang, J.; He, D.; Zhang, Q.; Pan, C.; Yuan, Q.; Shi, Y.; Tang, H.; Xu, F.; et al. ALDH2 (Aldehyde Dehydrogenase 2) Protects Against Hypoxia-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2303–2319. [Google Scholar] [CrossRef]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef]
- Palmer, K.R.; Jenkins, W.J. Impaired acetaldehyde oxidation in alcoholics. Gut 1982, 23, 729–733. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Cheung, C.; Smith, C.K.; Hoog, J.O.; Hotchkiss, S.A. Expression and localization of human alcohol and aldehyde dehydrogenase enzymes in skin. Biochem. Biophys. Res. Commun. 1999, 261, 100–107. [Google Scholar] [CrossRef]
- Ma, B.; Liu, Z.; Xu, H.; Liu, L.; Huang, T.; Meng, L.; Wang, L.; Zhang, Y.; Li, L.; Han, X. Molecular Characterization and Clinical Relevance of ALDH2 in Human Cancers. Front. Med. 2021, 8, 832605. [Google Scholar] [CrossRef]
- Yao, S.; Chen, W.; Zuo, H.; Bi, Z.; Zhang, X.; Pang, L.; Jing, Y.; Yin, X.; Cheng, H. Comprehensive Analysis of Aldehyde Dehydrogenases (ALDHs) and Its Significant Role in Hepatocellular Carcinoma. Biochem. Genet. 2022, 60, 1274–1297. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Abdelmegeed, M.A.; Yoo, S.H.; Kim, B.J.; Jo, S.A.; Jo, I.; Moon, K.H. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. J. Proteom. 2011, 74, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A. The Bidirectional Effect of Defective ALDH2 Polymorphism and Disease Prevention. Adv. Exp. Med. Biol. 2019, 1193, 69–87. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, L. The role of ALDH2 in tumorigenesis and tumor progression: Targeting ALDH2 as a potential cancer treatment. Acta Pharm. Sin. B 2021, 11, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The World of Melanoma: Epidemiologic, Genetic, and Anatomic Differences of Melanoma Across the Globe. Curr. Oncol. Rep. 2018, 20, 87. [Google Scholar] [CrossRef]
- Batta, N.; Shangraw, S.; Nicklawsky, A.; Yamauchi, T.; Zhai, Z.; Ravindran Menon, D.; Gao, D.; Dellavalle, R.P.; Fujita, M. Global melanoma correlations with obesity, smoking, and alcohol consumption. JMIR Dermatol. 2021, 4, e31275. [Google Scholar] [CrossRef]
- Li, K.; Guo, W.; Li, Z.; Wang, Y.; Sun, B.; Xu, D.; Ling, J.; Song, H.; Liao, Y.; Wang, T.; et al. ALDH2 Repression Promotes Lung Tumor Progression via Accumulated Acetaldehyde and DNA Damage. Neoplasia 2019, 21, 602–614. [Google Scholar] [CrossRef]
- Hou, G.; Chen, L.; Liu, G.; Li, L.; Yang, Y.; Yan, H.X.; Zhang, H.L.; Tang, J.; Yang, Y.C.; Lin, X.; et al. Aldehyde dehydrogenase-2 (ALDH2) opposes hepatocellular carcinoma progression by regulating AMP-activated protein kinase signaling in mice. Hepatology 2017, 65, 1628–1644. [Google Scholar] [CrossRef]
- Sule-Suso, J.; Pysanenko, A.; Spanel, P.; Smith, D. Quantification of acetaldehyde and carbon dioxide in the headspace of malignant and non-malignant lung cells in vitro by SIFT-MS. Analyst 2009, 134, 2419–2425. [Google Scholar] [CrossRef]
- Guan, X.; Rubin, E.; Anni, H. An optimized method for the measurement of acetaldehyde by high-performance liquid chromatography. Alcohol. Clin. Exp. Res. 2012, 36, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Umber, B.J.; Meinardi, S.; Leu, S.Y.; Zaldivar, F.; Blake, D.R.; Cooper, D.M. Acetaldehyde and hexanaldehyde from cultured white cells. J. Transl. Med. 2009, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Waris, S.; Patel, A.; Ali, A.; Mahmood, R. Acetaldehyde-induced oxidative modifications and morphological changes in isolated human erythrocytes: An in vitro study. Environ. Sci. Pollut. Res. Int. 2020, 27, 16268–16281. [Google Scholar] [CrossRef]
- Abdallah, M.A.; Singal, A.K. Mitochondrial dysfunction and alcohol-associated liver disease: A novel pathway and therapeutic target. Signal Transduct. Target. Ther. 2020, 5, 26. [Google Scholar] [CrossRef]
- Tsermpini, E.E.; Plemenitas Iljes, A.; Dolzan, V. Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review. Antioxidants 2022, 11, 1374. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhao, Z.; Sun, M.; Luo, J.; Xiao, Y. ALDH2 gene polymorphism in different types of cancers and its clinical significance. Life Sci. 2016, 147, 59–66. [Google Scholar] [CrossRef]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. U S A 2021, 118, e2000915118. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Yarla, N.; Haykal, T.; Nguyen, Y.V.; Cao, L.; Ferreira, M.; Holtzhausen, A.; Al-Rohil, R.; Salama, A.K.S.; Beasley, G.M.; et al. Tumor-intrinsic NLRP3-HSP70-TLR4 axis drives premetastatic niche development and hyperprogression during anti-PD-1 immunotherapy. Sci. Transl. Med. 2022, 14, eabq7019. [Google Scholar] [CrossRef]
- Endo, J.; Sano, M.; Katayama, T.; Hishiki, T.; Shinmura, K.; Morizane, S.; Matsuhashi, T.; Katsumata, Y.; Zhang, Y.; Ito, H.; et al. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ. Res. 2009, 105, 1118–1127. [Google Scholar] [CrossRef]
- Chauhan, J.S.; Holzel, M.; Lambert, J.P.; Buffa, F.M.; Goding, C.R. The MITF regulatory network in melanoma. Pigment. Cell Melanoma Res. 2022, 35, 517–533. [Google Scholar] [CrossRef]
- Shortall, K.; Djeghader, A.; Magner, E.; Soulimane, T. Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective. Front. Mol. Biosci. 2021, 8, 659550. [Google Scholar] [CrossRef]
- Singh, S.; Arcaroli, J.; Thompson, D.C.; Messersmith, W.; Vasiliou, V. Acetaldehyde and retinaldehyde-metabolizing enzymes in colon and pancreatic cancers. Adv. Exp. Med. Biol. 2015, 815, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Li, J.; Donini, S.; Sobol, R.W.; Rizzi, M.; Garavaglia, S. Crystal structure of human aldehyde dehydrogenase 1A3 complexed with NAD+ and retinoic acid. Sci. Rep. 2016, 6, 35710. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Simonis, V.; Del Gaudio, C.; Cucini, C.; Ziche, M.; Morbidelli, L.; Donnini, S. ALDH1A1 confers resistance to RAF/MEK inhibitors in melanoma cells by maintaining stemness phenotype and activating PI3K/AKT signaling. Biochem. Pharmacol. 2024, 224, 116252. [Google Scholar] [CrossRef]
- Lavudi, K.; Nuguri, S.M.; Pandey, P.; Kokkanti, R.R.; Wang, Q.E. ALDH and cancer stem cells: Pathways, challenges, and future directions in targeted therapy. Life Sci. 2024, 356, 123033. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Yang, Y.; Qiu, Y.; Wang, Z.; Li, X.; Zhang, W. Emerging roles of activating transcription factor (ATF) family members in tumourigenesis and immunity: Implications in cancer immunotherapy. Genes. Dis. 2022, 9, 981–999. [Google Scholar] [CrossRef]
- Wei, P.L.; Prince, G.; Batzorig, U.; Huang, C.Y.; Chang, Y.J. ALDH2 promotes cancer stemness and metastasis in colorectal cancer through activating beta-catenin signaling. J. Cell Biochem. 2023, 124, 907–920. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, Z.; Yamauchi, T.; Sandoval, K.; Villarreal, K.; Kwong, M.W.C.; Swanson, E.J.; Tan, A.C.; Fujita, M. Downregulated ALDH2 Contributes to Tumor Progression and Targeted Therapy Resistance in Human Metastatic Melanoma Cells. Cells 2025, 14, 913. https://doi.org/10.3390/cells14120913
Zhai Z, Yamauchi T, Sandoval K, Villarreal K, Kwong MWC, Swanson EJ, Tan AC, Fujita M. Downregulated ALDH2 Contributes to Tumor Progression and Targeted Therapy Resistance in Human Metastatic Melanoma Cells. Cells. 2025; 14(12):913. https://doi.org/10.3390/cells14120913
Chicago/Turabian StyleZhai, Zili, Takeshi Yamauchi, Karenna Sandoval, Kira Villarreal, Man Wai Charlotte Kwong, Emily J. Swanson, Aik Choon Tan, and Mayumi Fujita. 2025. "Downregulated ALDH2 Contributes to Tumor Progression and Targeted Therapy Resistance in Human Metastatic Melanoma Cells" Cells 14, no. 12: 913. https://doi.org/10.3390/cells14120913
APA StyleZhai, Z., Yamauchi, T., Sandoval, K., Villarreal, K., Kwong, M. W. C., Swanson, E. J., Tan, A. C., & Fujita, M. (2025). Downregulated ALDH2 Contributes to Tumor Progression and Targeted Therapy Resistance in Human Metastatic Melanoma Cells. Cells, 14(12), 913. https://doi.org/10.3390/cells14120913