
Long-Term Treatment with Low-Level Arsenite Induces Aberrant Proliferation and Migration via Redox Rebalance in Human Urothelial Cells


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Different Reagents
2.2. Cell Culture and Treatment
2.3. Cell Viability Assay
2.4. Assessment of Intracellular ROS Levels
2.5. Evaluation of TrxR1 and GR Enzymatic Activity Together with Intracellular GSH Levels
2.6. Transwell Assay
2.7. Wound Healing Assay
2.8. Western Blot Analysis
2.9. Animals and Treatments
2.10. ELISA Test
2.11. Immunofluorescence Staining
2.12. Statistical Analysis
3. Results
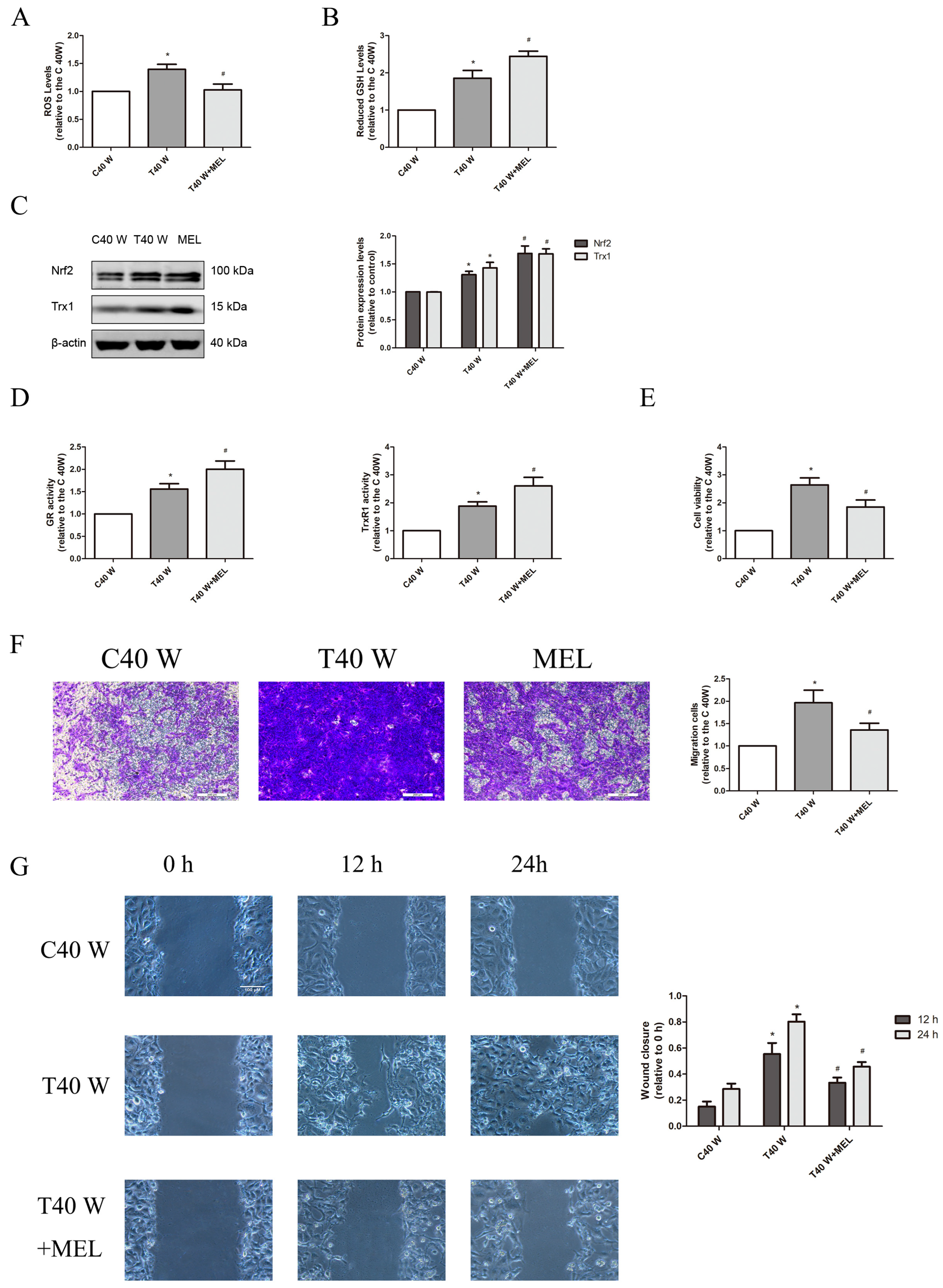
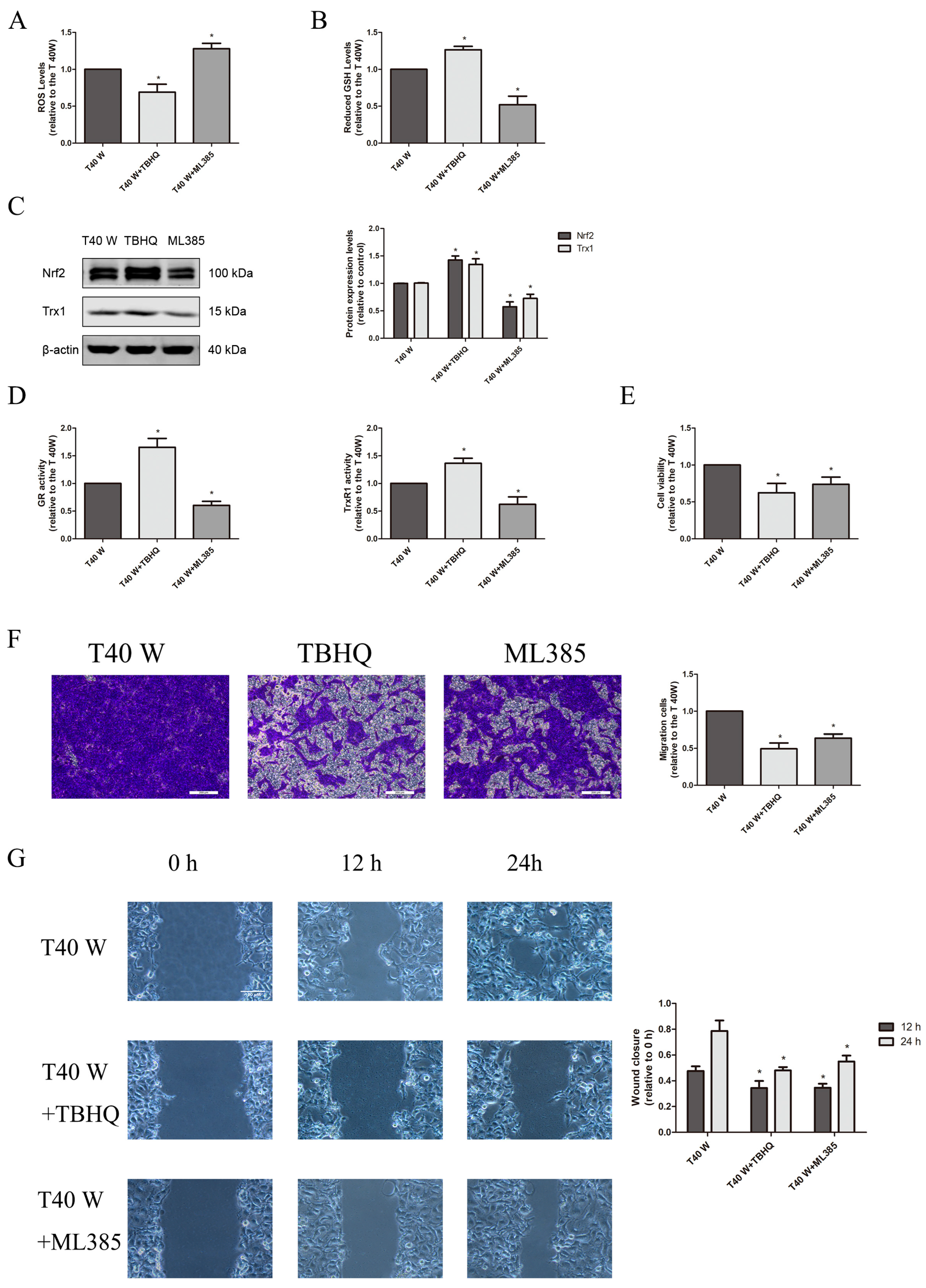
3.1. Alteration of Redox Control Systems and Their Roles in the Proliferation and Migration of SV-HUC-1 Cells Following Extended Arsenite Exposure
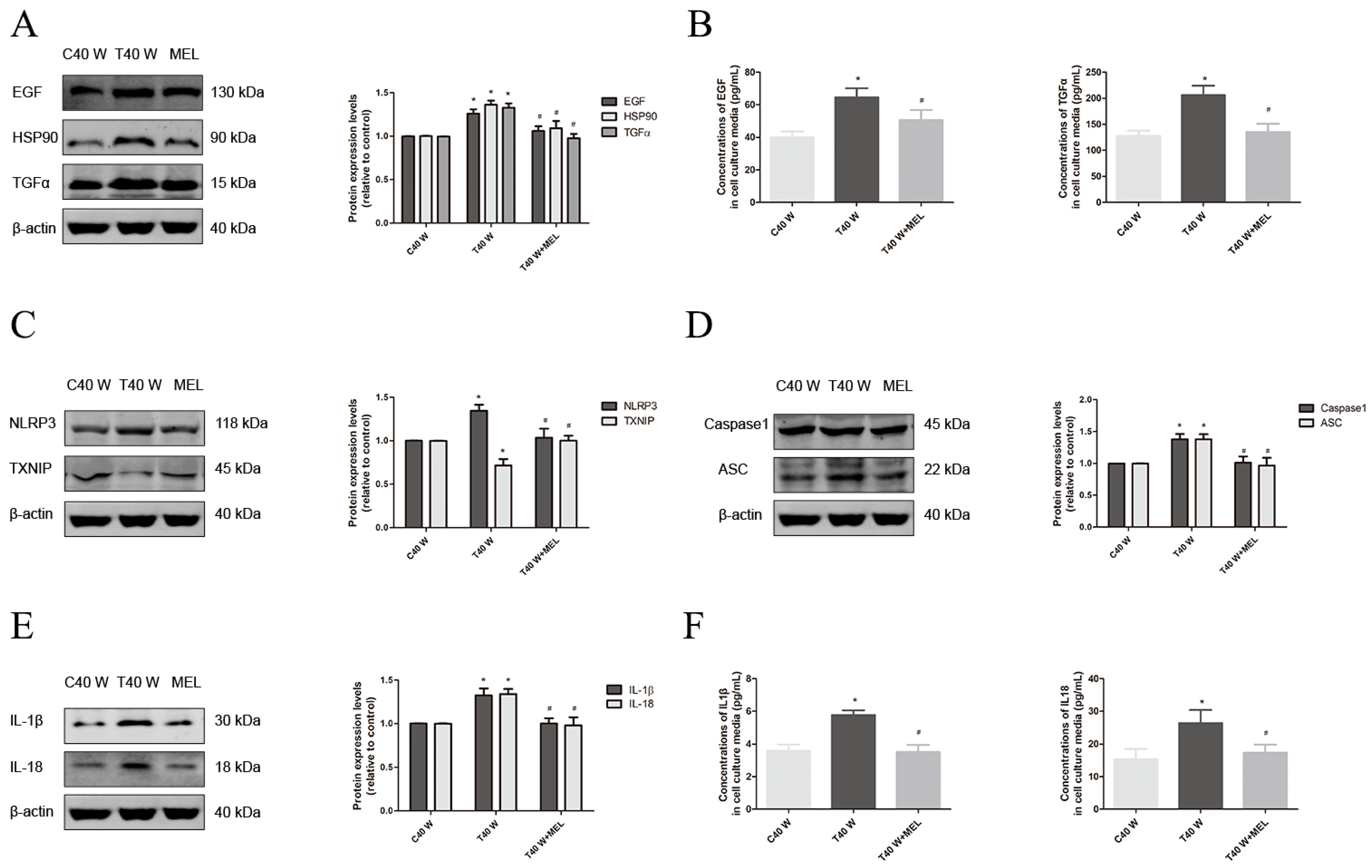
3.2. Mechanisms of EGF, TGFα, and HSP90 Overexpression, as Well as Aberrant Cell Proliferation and Migration, in SV-HUC-1 Cells Following Extended Arsenite Treatment
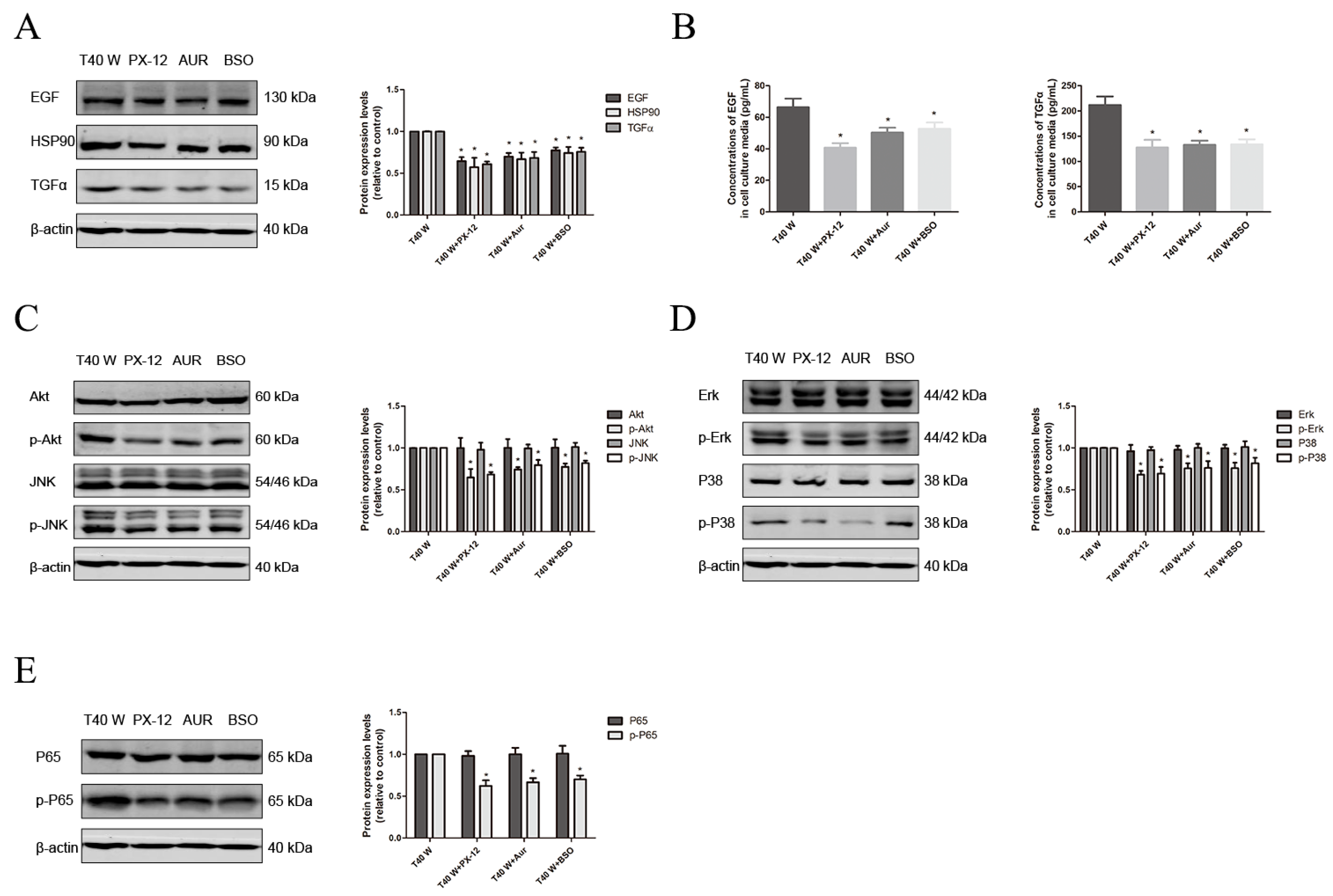
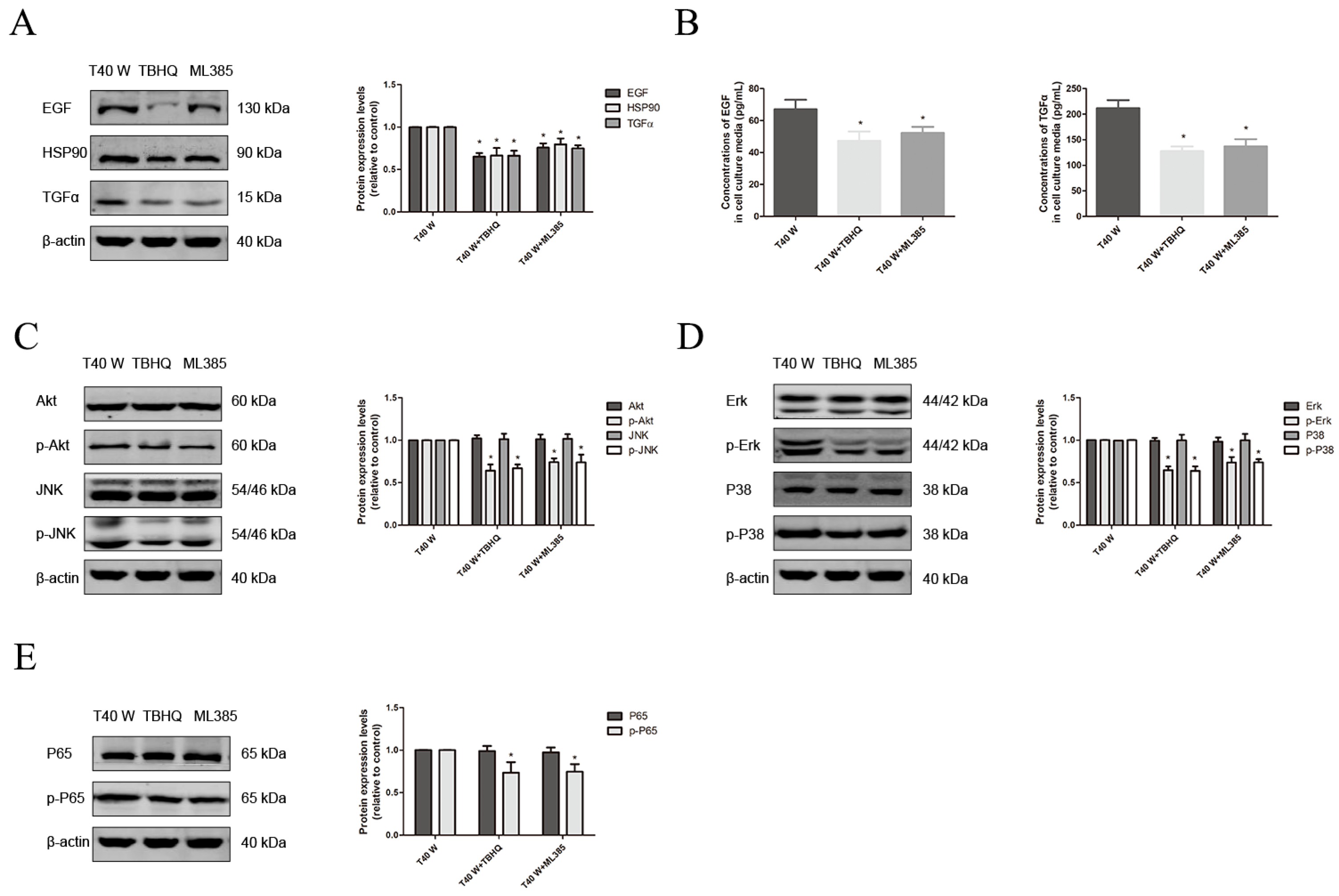
3.3. Effects of the NLRP3 Inflammasome on Cell Proliferation and Migration, Enhancement of HSP90, TGFα and EGF Levels, and Activation of Related Signaling Pathways Following Long-Term Arsenite Treatment
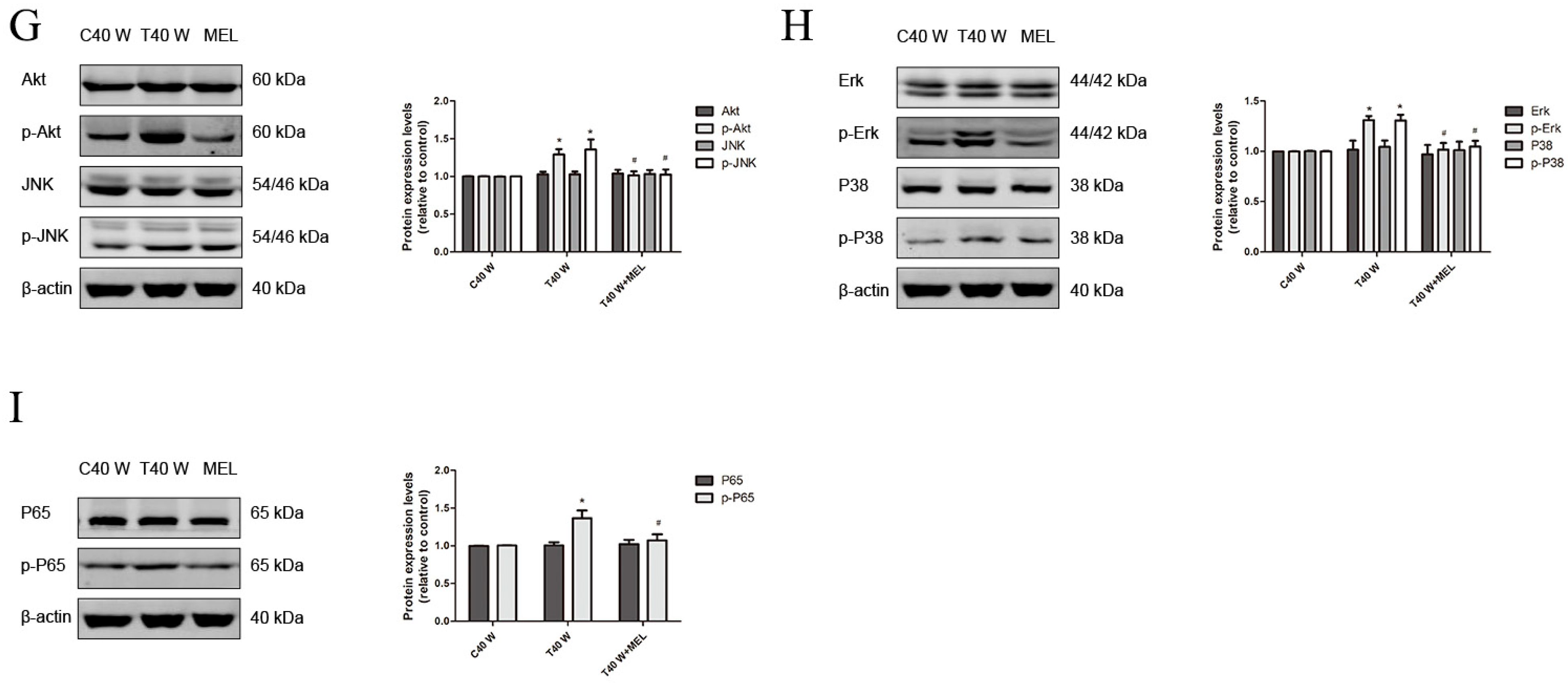
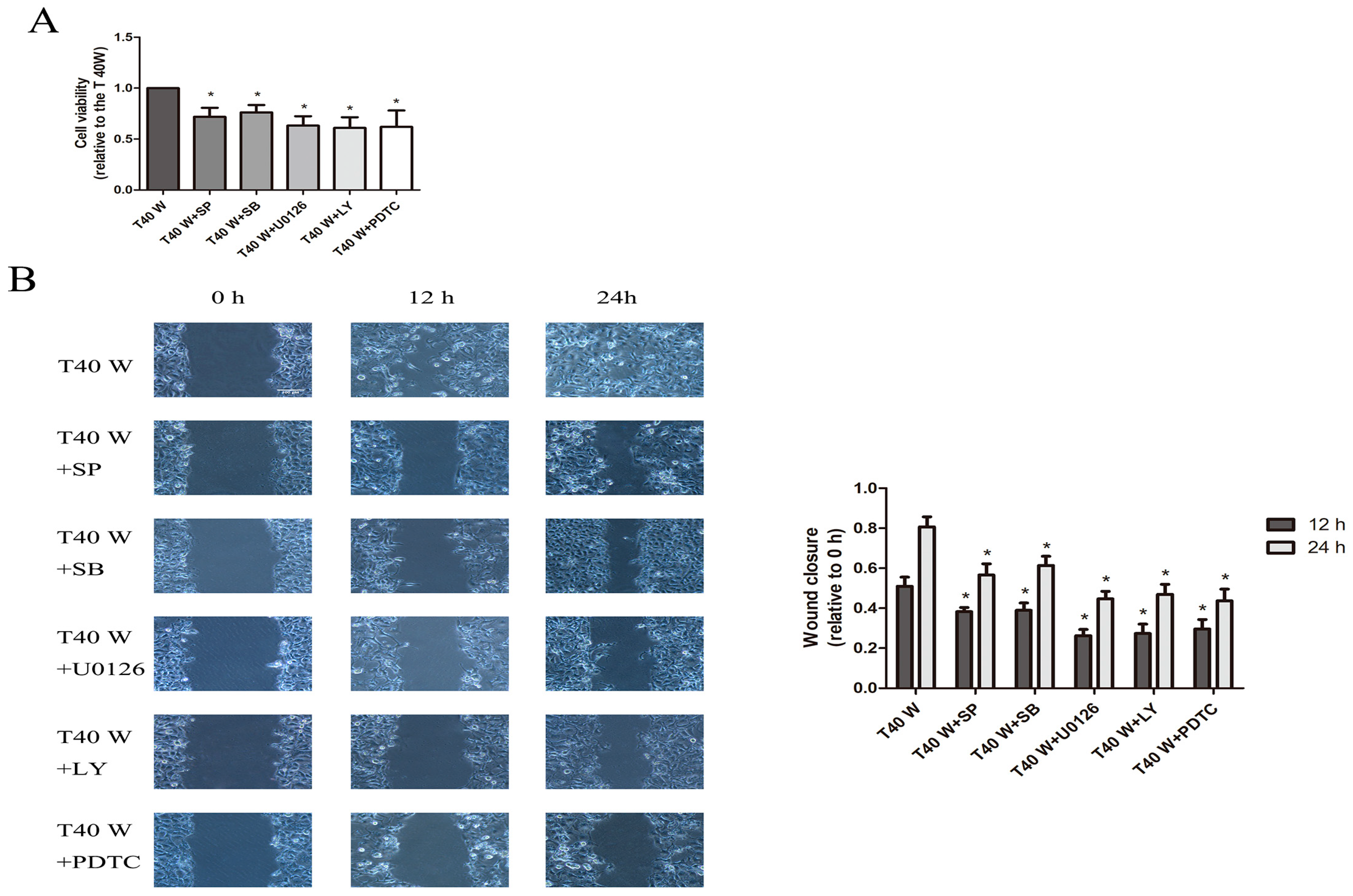
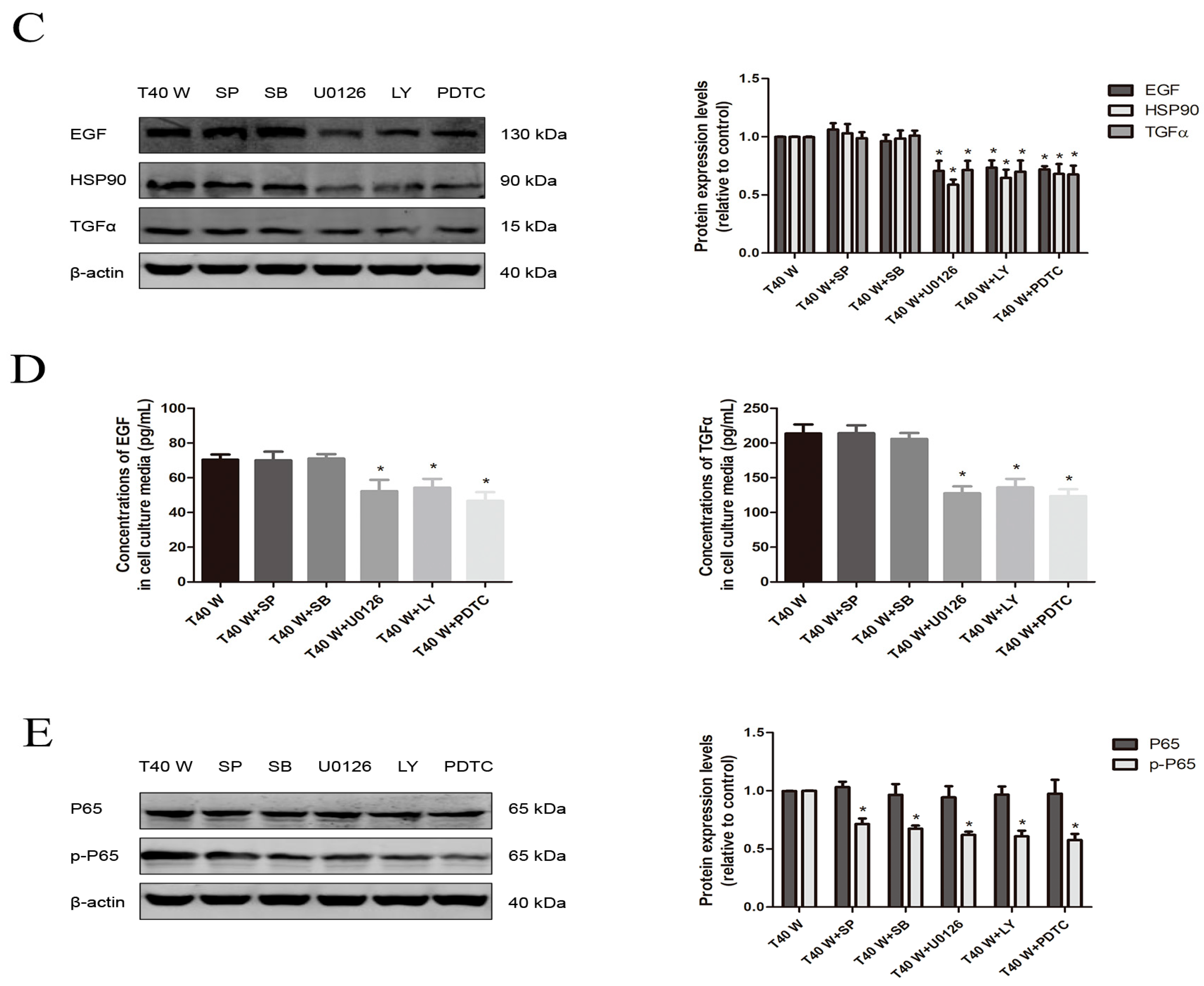
3.4. Effects of JNK, p38, ERK, AKT, or NF-κB on Cell Proliferation and Migration and the Enhancement of HSP90, TGFα, and EGF Following Long-Term Arsenite Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EGF | epidermal growth factor |
| Grx | glutaredoxin |
| GR | glutathione reductase |
| GSH | reduced glutathione |
| HSP90 | heat shock protein 90 |
| NLRP3 | Nod-like receptor protein 3 |
| TGFα | transforming growth factor α |
| Trx | thioredoxin |
| TrxR | Trx reductase |
| TXNIP | Thioredoxin-interacting protein |
References
- Di Giovanni, P.; Di Martino, G.; Scampoli, P.; Cedrone, F.; Meo, F.; Lucisano, G.; Romano, F.; Staniscia, T. Arsenic Exposure and Risk of Urothelial Cancer: Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 3105. [Google Scholar] [CrossRef]
- Wei, M.; Wanibuchi, H.; Yamamoto, S.; Li, W.; Fukushima, S. Urinary bladder carcinogenicity of dimethylarsinic acid in male F344 rats. Carcinogenesis 1999, 20, 1873–1876. [Google Scholar] [CrossRef]
- Jin, P.; Liu, J.; Zhou, Q.; Li, S.; Liu, W.; Xi, S. Long-term treatment with arsenite activates HER1 and HER2 through upregulating EGF, TGFalpha, and HSP90 in a human uroepithelial cell line. Cell Biol. Toxicol. 2020, 36, 279–284. [Google Scholar] [CrossRef]
- Zhou, Q.; Jin, P.; Liu, J.; Wang, F.; Xi, S. HER2 and Src co-regulate proliferation, migration and transformation by downstream signaling pathways in arsenite-treated human uroepithelial cells. Met. Integr. Biometal Sci. 2018, 10, 1141–1159. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Investig. 2016, 126, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [PubMed]
- Fath, M.A.; Ahmad, I.M.; Smith, C.J.; Spence, J.; Spitz, D.R. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2011, 17, 6206–6217. [Google Scholar] [CrossRef]
- Scarbrough, P.M.; Mapuskar, K.A.; Mattson, D.M.; Gius, D.; Watson, W.H.; Spitz, D.R. Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Free Radic. Biol. Med. 2012, 52, 436–443. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules—From biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, D.F.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Mandal, P.K.; Schneider, M.; Kolle, P.; Kuhlencordt, P.; Forster, H.; Beck, H.; Bornkamm, G.W.; Conrad, M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010, 70, 9505–9514. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid. Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat. Res. 2016, 186, 385–395. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Zhao, L.; Wei, Y.; Feng, H.; Wang, C.; Wei, W.; Ding, Y.; Sun, D. Changes in serum thioredoxin among individuals chronically exposed to arsenic in drinking water. Toxicol. Appl. Pharmacol. 2012, 259, 124–132. [Google Scholar] [CrossRef]
- Hu, Y.; Jin, X.; Snow, E.T. Effect of arsenic on transcription factor AP-1 and NF-kappaB DNA binding activity and related gene expression. Toxicol. Lett. 2002, 133, 33–45. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Gao, Y.; Zhang, W.; Cui, X.; Liu, J.; Wei, Y. Arsenic Induces Thioredoxin 1 and Apoptosis in Human Liver HHL-5 Cells. Biol. Trace Elem. Res. 2018, 181, 234–241. [Google Scholar] [CrossRef]
- Jin, P.; Zhou, Q.; Xi, S. Low-dose arsenite causes overexpression of EGF, TGFalpha, and HSP90 through Trx1-TXNIP-NLRP3 axis mediated signaling pathways in the human bladder epithelial cells. Ecotoxicol. Environ. Saf. 2022, 247, 114263. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Liu, J.; Wang, X.; Yang, L.; Zhou, Q.; Lin, X.; Xi, S. HER2 Activation Factors in Arsenite-Exposed Bladder Epithelial Cells. Toxicol. Sci. Off. J. Soc. Toxicol. 2018, 166, 354–369. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Zhu, X.; Huang, C.; Peng, B. Overexpression of thioredoxin system proteins predicts poor prognosis in patients with squamous cell carcinoma of the tongue. Oral. Oncol. 2011, 47, 609–614. [Google Scholar] [CrossRef]
- Lin, F.; Zhang, P.; Zuo, Z.; Wang, F.; Bi, R.; Shang, W.; Wu, A.; Ye, J.; Li, S.; Sun, X.; et al. Thioredoxin-1 promotes colorectal cancer invasion and metastasis through crosstalk with S100P. Cancer Lett. 2017, 401, 1–10. [Google Scholar] [CrossRef]
- You, B.R.; Shin, H.R.; Park, W.H. PX-12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS-dependent apoptosis. Int. J. Oncol. 2014, 44, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; You, B.R.; Park, W.H. PX-12-induced HeLa cell death is associated with oxidative stress and GSH depletion. Oncol. Lett. 2013, 6, 1804–1810. [Google Scholar] [CrossRef]
- Wang, F.; Lin, F.; Zhang, P.; Ni, W.; Bi, L.; Wu, J.; Jiang, L. Thioredoxin-1 inhibitor, 1-methylpropyl 2-imidazolyl disulfide, inhibits the growth, migration and invasion of colorectal cancer cell lines. Oncol. Rep. 2015, 33, 967–973. [Google Scholar] [CrossRef]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Zhang, H.; Rose, B.J.; Pyuen, A.A.; Thamm, D.H. In vitro antineoplastic effects of auranofin in canine lymphoma cells. BMC Cancer 2018, 18, 522. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. BCR 2010, 12, R44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, Q.; Chng, W.J. TXNIP (VDUP-1, TBP-2): A major redox regulator commonly suppressed in cancer by epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2011, 43, 1668–1673. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, S.H.; Woo, G.H.; Kwon, H.J.; Kim, D.Y. Downregulation of TXNIP leads to high proliferative activity and estrogen-dependent cell growth in breast cancer. Biochem. Biophys. Res. Commun. 2018, 498, 566–572. [Google Scholar] [CrossRef]
- Nishizawa, K.; Nishiyama, H.; Matsui, Y.; Kobayashi, T.; Saito, R.; Kotani, H.; Masutani, H.; Oishi, S.; Toda, Y.; Fujii, N.; et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis 2011, 32, 1459–1466. [Google Scholar] [CrossRef]
- Li, J.; Yue, Z.; Xiong, W.; Sun, P.; You, K.; Wang, J. TXNIP overexpression suppresses proliferation and induces apoptosis in SMMC7221 cells through ROS generation and MAPK pathway activation. Oncol. Rep. 2017, 37, 3369–3376. [Google Scholar] [CrossRef]
- Eblin, K.E.; Bredfeldt, T.G.; Buffington, S.; Gandolfi, A.J. Mitogenic signal transduction caused by monomethylarsonous acid in human bladder cells: Role in arsenic-induced carcinogenesis. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 95, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hock, J.M.; Sullivan, C.; Fang, G.; Cox, A.J.; Davis, K.T.; Davis, B.H.; Li, X. Activation of the p38 MAPK/Akt/ERK1/2 signal pathways is required for the protein stabilization of CDC6 and cyclin D1 in low-dose arsenite-induced cell proliferation. J. Cell. Biochem. 2010, 111, 1546–1555. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Jiang, Y.; Jing, Y.; He, J.; Rojanasakul, Y.; Liu, L.Z.; Jiang, B.H. Arsenite induces cell transformation by reactive oxygen species, AKT, ERK1/2, and p70S6K1. Biochem. Biophys. Res. Commun. 2011, 414, 533–538. [Google Scholar] [CrossRef]
- Ling, M.; Li, Y.; Xu, Y.; Pang, Y.; Shen, L.; Jiang, R.; Zhao, Y.; Yang, X.; Zhang, J.; Zhou, J.; et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-kappaB in arsenite-induced cell transformation. Free Radic. Biol. Med. 2012, 52, 1508–1518. [Google Scholar] [CrossRef]
- Liu, S.; Wang, F.; Yan, L.; Zhang, L.; Song, Y.; Xi, S.; Jia, J.; Sun, G. Oxidative stress and MAPK involved into ATF2 expression in immortalized human urothelial cells treated by arsenic. Arch. Toxicol. 2013, 87, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Sartelet, H.; Rougemont, A.L.; Fabre, M.; Castaing, M.; Duval, M.; Fetni, R.; Michiels, S.; Beaunoyer, M.; Vassal, G. Activation of the phosphatidylinositol 3′-kinase/AKT pathway in neuroblastoma and its regulation by thioredoxin 1. Hum. Human Pathol. 2011, 42, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.Y.; Lee, S.W.; Shin, Y.H.; Lee, J.H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Zhou, Q.; Xi, S.; Jin, P. Long-Term Treatment with Low-Level Arsenite Induces Aberrant Proliferation and Migration via Redox Rebalance in Human Urothelial Cells. Cells 2025, 14, 912. https://doi.org/10.3390/cells14120912
Yan X, Zhou Q, Xi S, Jin P. Long-Term Treatment with Low-Level Arsenite Induces Aberrant Proliferation and Migration via Redox Rebalance in Human Urothelial Cells. Cells. 2025; 14(12):912. https://doi.org/10.3390/cells14120912
Chicago/Turabian StyleYan, Xiangli, Qing Zhou, Shuhua Xi, and Peiyu Jin. 2025. "Long-Term Treatment with Low-Level Arsenite Induces Aberrant Proliferation and Migration via Redox Rebalance in Human Urothelial Cells" Cells 14, no. 12: 912. https://doi.org/10.3390/cells14120912
APA StyleYan, X., Zhou, Q., Xi, S., & Jin, P. (2025). Long-Term Treatment with Low-Level Arsenite Induces Aberrant Proliferation and Migration via Redox Rebalance in Human Urothelial Cells. Cells, 14(12), 912. https://doi.org/10.3390/cells14120912