
Chimeric Antigen Receptor (CAR) T Cells Releasing Soluble SLAMF6 Isoform 2 Gain Superior Anti-Cancer Cell Functionality in an Auto-Stimulatory Fashion

 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. CAR T Cell Generation
2.3. Flow Cytometry
2.4. Cytokine Secretion
2.5. Cytotoxicity Assay
2.6. Repetitive Stimulation Assay
2.7. Statistical Analysis
3. Results
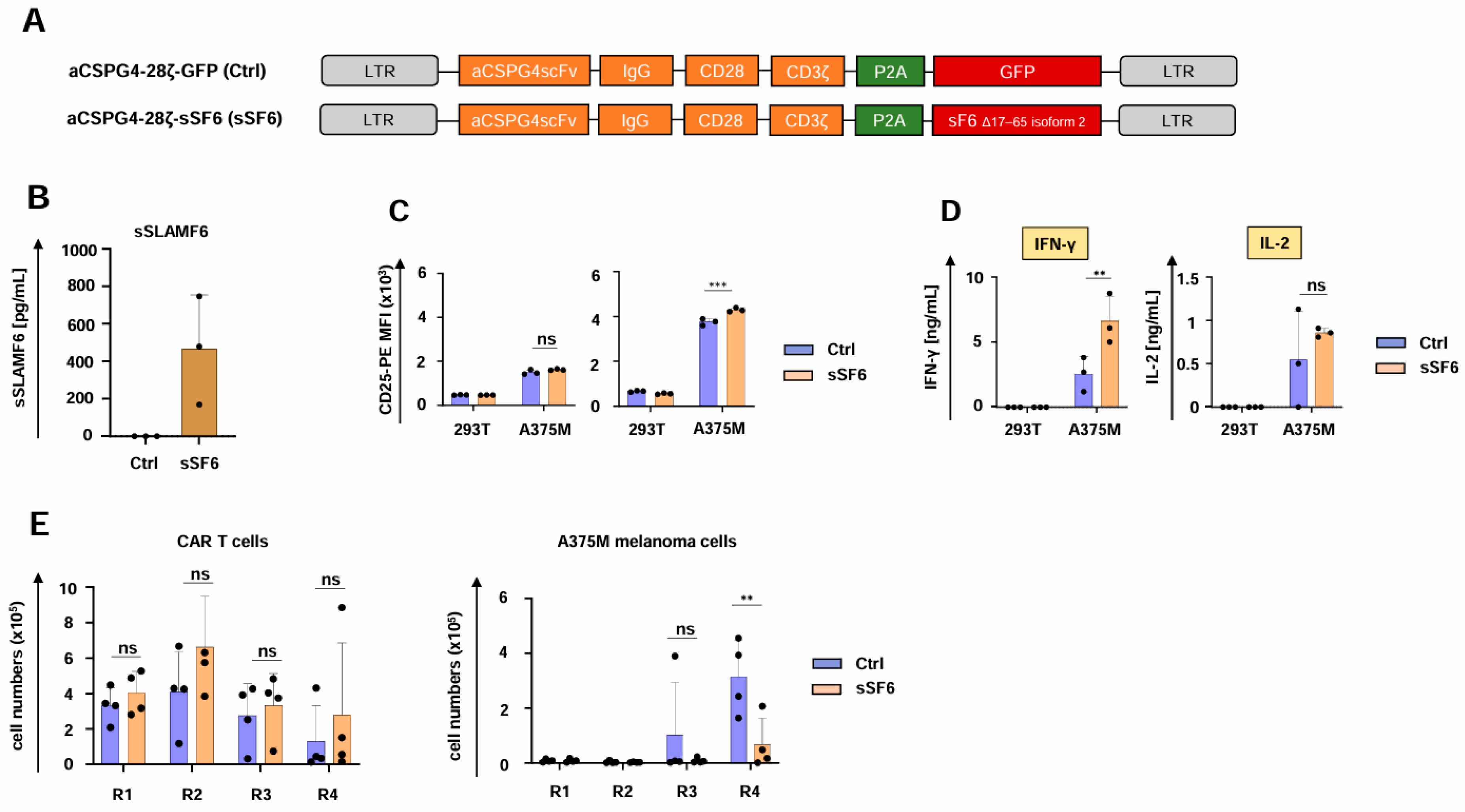
3.1. Design of CAR T Cells Releasing Soluble SLAMF6 Isoform 2
3.2. CAR T Cells Releasing Soluble SLAMF6 Isoform 2 Display Increased IFN-γ Secretion
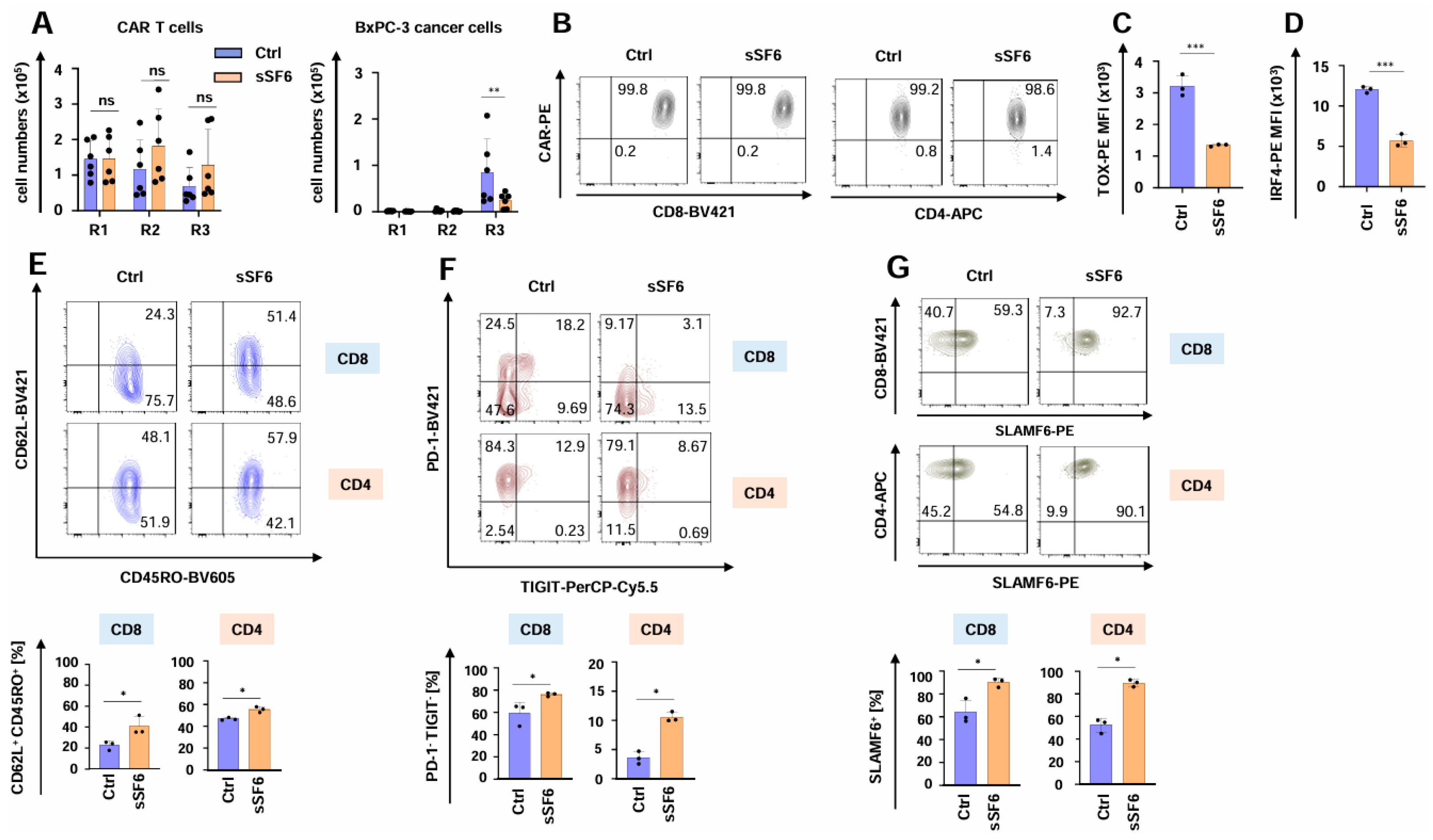
3.3. CAR T Cells Releasing Soluble SLAMF6 Isoform 2 Show Augmented Functional Persistence Under Repetitive Stimulatory Conditions
3.4. CAR T Cells Releasing Soluble SLAMF6 Isoform 2 Exhibit Enhanced Functionality Against Melanoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BLIMP-1 | B-lymphocyte-induced maturation protein 1 |
| CAR | Chimeric antigen receptor |
| CD | Cluster of differentiation |
| CSPG4 | Chondroitin Sulfate Proteoglycan 4 |
| CEA | Carcinoembryonic antigen |
| ELISA | Enzyme-linked Immunosorbent Assay |
| GFP | Green fluorescent protein |
| gp100 | Glycoprotein 100 |
| IL | Interleukin |
| IFN-γ | Interferon-Gamma |
| IRF4 | Interferon Regulatory Factor 4 |
| LAG3 | Lymphocyte-activation gene 3 |
| mAb | Monoclonal antibody |
| NK cells | Natural killer cells |
| PBMCs | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death protein 1 |
| scFv | Single-chain variable Fragment |
| SLAMF6 | SLAM family member 6 |
| TCF-1 | T cell factor 1 |
| TIGIT | T-cell immunoreceptor with immunoglobulin and ITIM domains |
| TIM3 | T cell immunoglobulin mucin-3 |
| TOX | Thymocyte selection-associated high mobility group box protein |
References
- Uslu, U.; June, C.H. Beyond the blood: Expanding CAR T cell therapy to solid tumors. Nat. Biotechnol. 2025, 43, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Dabas, P.; Danda, A. Revolutionizing cancer treatment: A comprehensive review of CAR-T cell therapy. Med. Oncol. 2023, 40, 275. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; Castelli, S.; June, C.H. CAR T cell combination therapies to treat cancer. Cancer Cell 2024, 42, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Kembuan, G.J.; Kim, J.Y.; Maus, M.V.; Jan, M. Targeting solid tumor antigens with chimeric receptors: Cancer biology meets synthetic immunology. Trends Cancer 2024, 10, 312–331. [Google Scholar] [CrossRef]
- Posey, A.D.; Young, R.M.; June, C.H. Future perspectives on engineered T cells for cancer. Trends Cancer 2024, 10, 687–695. [Google Scholar] [CrossRef]
- Noll, J.H.; Levine, B.L.; June, C.H.; Fraietta, J.A. Beyond youth: Understanding CAR T cell fitness in the context of immunological aging. Semin. Immunol. 2023, 70, 101840. [Google Scholar] [CrossRef]
- Singh, N.; Maus, M.V. Synthetic manipulation of the cancer-immunity cycle: CAR-T cell therapy. Immunity 2023, 56, 2296–2310. [Google Scholar] [CrossRef] [PubMed]
- Kronig, M.N.; Wehrli, M.; Salas-Benito, D.; Maus, M.V. Hurdles race for CAR T-cell therapy in digestive tract cancer. Immunol. Rev. 2023, 320, 100–119. [Google Scholar] [CrossRef]
- Maus, M.V. Signatures of dysfunctional CAR T cells. Blood 2023, 141, 3127–3129. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648, Erratum in Nature 2023, 619, E26. https://doi.org/10.1038/s41586-023-06088-3. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lee, H.J.; Kim, H.C.; Lee, Y.; Nam, S.K.; Hupperetz, C.; Ma, J.S.; Wang, X.; Singer, O.; Kim, W.S.; et al. PD-1 and TIGIT downregulation distinctly affect the effector and early memory phenotypes of CD19-targeting CAR T cells. Mol. Ther. 2022, 30, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Jafarzadeh, L.; Masoumi, E.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Khakpoor-Koosheh, M.; Rostamian, H.; Noorbakhsh, F.; Hadjati, J. Targeted knockdown of Tim3 by short hairpin RNAs improves the function of anti-mesothelin CAR T cells. Mol. Immunol. 2021, 139, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef]
- Harrer, D.C.; Bezler, V.; Hartley, J.; Herr, W.; Abken, H. IRF4 downregulation improves sensitivity and endurance of CAR T cell functional capacities. Front. Immunol. 2023, 14, 1185618. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.Y.; Narayan, V.; McDonald, S.; Rech, A.J.; Bartoszek, R.; Hong, G.; Davis, M.M.; Xu, J.; Boesteanu, A.C.; Barber-Rotenberg, J.S.; et al. BLIMP1 and NR4A3 transcription factors reciprocally regulate antitumor CAR T cell stemness and exhaustion. Sci. Transl. Med. 2022, 14, eabn7336. [Google Scholar] [CrossRef]
- Doan, A.E.; Mueller, K.P.; Chen, A.Y.; Rouin, G.T.; Chen, Y.; Daniel, B.; Lattin, J.; Markovska, M.; Mozarsky, B.; Arias-Umana, J.; et al. FOXO1 is a master regulator of memory programming in CAR T cells. Nature 2024, 629, 211–218. [Google Scholar] [CrossRef]
- Gautam, S.; Fioravanti, J.; Zhu, W.; Le Gall, J.B.; Brohawn, P.; Lacey, N.E.; Hu, J.; Hocker, J.D.; Hawk, N.V.; Kapoor, V.; et al. The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nat. Immunol. 2019, 20, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef]
- Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells 2020, 9, 873. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Gunes, M.; Rosen, S.T.; Shachar, I.; Gunes, E.G. Signaling lymphocytic activation molecule family receptors as potential immune therapeutic targets in solid tumors. Front. Immunol. 2024, 15, 1297473. [Google Scholar] [CrossRef]
- Eisenberg, G.; Engelstein, R.; Geiger-Maor, A.; Hajaj, E.; Merims, S.; Frankenburg, S.; Uzana, R.; Rutenberg, A.; Machlenkin, A.; Frei, G.; et al. Soluble SLAMF6 Receptor Induces Strong CD8+ T-cell Effector Function and Improves Anti-Melanoma Activity In Vivo. Cancer Immunol. Res. 2018, 6, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Hajaj, E.; Zisman, E.; Tzaban, S.; Merims, S.; Cohen, J.; Klein, S.; Frankenburg, S.; Sade-Feldman, M.; Tabach, Y.; Yizhak, K.; et al. Alternative Splicing of the Inhibitory Immune Checkpoint Receptor SLAMF6 Generates a Dominant Positive Form, Boosting T-cell Effector Functions. Cancer Immunol. Res. 2021, 9, 637–650. [Google Scholar] [CrossRef]
- Harrer, D.C.; Eder, M.; Barden, M.; Pan, H.; Herr, W.; Abken, H. Ectopic PU.1 Expression Provides Chimeric Antigen Receptor (CAR) T Cells with Innate Cell Capacities Including IFN-β Release. Cancers 2024, 16, 2737. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Rappl, G.; AbkenH. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T Cell Response against CD30− Tumors. Mol. Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef]
- Harrer, D.C.; Schenkel, C.; Bezler, V.; Kaljanac, M.; Hartley, J.; Barden, M.; Pan, H.; Holzinger, A.; Herr, W.; Abken, H. CAR Triggered Release of Type-1 Interferon Limits CAR T-Cell Activities by an Artificial Negative Autocrine Loop. Cells 2022, 11, 3839. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Dragovich, M.A.; Adam, K.; Strazza, M.; Tocheva, A.S.; Peled, M.; Mor, A. SLAMF6 clustering is required to augment T cell activation. PLoS ONE 2019, 14, e0218109. [Google Scholar] [CrossRef]
- Oba, T.; Long, M.D.; Ito, K.I.; Ito, F. Clinical and immunological relevance of SLAMF6 expression in the tumor microenvironment of breast cancer and melanoma. Sci. Rep. 2024, 14, 2394. [Google Scholar] [CrossRef]
- Goodman, D.B.; Azimi, C.S.; Kearns, K.; Talbot, A.; Garakani, K.; Garcia, J.; Patel, N.; Hwang, B.; Lee, D.; Park, E.; et al. Pooled screening of CAR T cells identifies diverse immune signaling domains for next-generation immunotherapies. Sci. Transl. Med. 2022, 14, eabm1463. [Google Scholar] [CrossRef]
- Yigit, B.; Wang, N.; Ten Hacken, E.; Chen, S.S.; Bhan, A.K.; Suarez-Fueyo, A.; Katsuyama, E.; Tsokos, G.C.; Chiorazzi, N.; Wu, C.J.; et al. SLAMF6 as a Regulator of Exhausted CD8+ T Cells in Cancer. Cancer Immunol. Res. 2019, 7, 1485–1496. [Google Scholar] [CrossRef]
- Hajaj, E.; Eisenberg, G.; Klein, S.; Frankenburg, S.; Merims, S.; Ben David, I.; Eisenhaure, T.; Henrickson, S.E.; Villani, A.C.; Hacohen, N.; et al. SLAMF6 deficiency augments tumor killing and skews toward an effector phenotype revealing it as a novel T cell checkpoint. eLife 2020, 9, e52539. [Google Scholar] [CrossRef]
- Banta, K.L.; Xu, X.; Chitre, A.S.; Au-Yeung, A.; Takahashi, C.; O’Gorman, W.E.; Wu, T.D.; Mittman, S.; Cubas, R.; Comps-Agrar, L.; et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity 2022, 55, 512–526.e9. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Graham, C.E.; Berger, T.R.; Ritchey, J.; Horick, N.K.; El-Jawahri, A.; Scarfò, I.; Schmidts, A.; Haradhvala, N.J.; Wehrli, M.; et al. Phase 1 study of CAR-37 T cells in patients with relapsed or refractory CD37+ lymphoid malignancies. Blood 2024, 144, 1153–1167. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Isaacson, J.; Bhanap, P.; Putnam, N.; Padilla, J.; Fatima, N.; Dotson, M.; Hayoun, D.; Ahmadi, M.; Nonterah, G.; Ji, Y. Enhancing CAR T-cell therapy manufacturing efficiency through semi-automated bioprocessing. Clin. Transl. Immunol. 2025, 14, e70025. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schelker, R.C.; Fioravanti, J.; Mastrogiovanni, F.; Baldwin, J.G.; Rana, N.; Li, P.; Chen, P.; Vadász, T.; Spolski, R.; Heuser-Loy, C.; et al. LIM-domain-only 4 (LMO4) enhances CD8+ T-cell stemness and tumor rejection by boosting IL-21-STAT3 signaling. Signal Transduct. Target. Ther. 2024, 9, 199. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sato, N.; Sabzevari, H.; Fu, S.; Ju, W.; Petrus, M.N.; Bamford, R.N.; Waldmann, T.A.; Tagaya, Y. Development of an IL-15-autocrine CD8 T-cell leukemia in IL-15-transgenic mice requires the cis expression of IL-15Rα. Blood 2011, 117, 4032–4040. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Svoboda, J.; Landsburg, D.J.; Gerson, J.; Nasta, S.D.; Barta, S.K.; Chong, E.A.; Cook, M.; Frey, N.V.; Shea, J.; Cervini, A.; et al. Enhanced CAR T-Cell Therapy for Lymphoma after Previous Failure. N. Engl. J. Med. 2025, 392, 1824–1835. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Schlierkamp-Voosen, T.; Barden, M.; Pan, H.; Xydia, M.; Herr, W.; Dörrie, J.; Schaft, N.; Abken, H. Chimeric Antigen Receptor (CAR) T Cells Releasing Soluble SLAMF6 Isoform 2 Gain Superior Anti-Cancer Cell Functionality in an Auto-Stimulatory Fashion. Cells 2025, 14, 901. https://doi.org/10.3390/cells14120901
Harrer DC, Schlierkamp-Voosen T, Barden M, Pan H, Xydia M, Herr W, Dörrie J, Schaft N, Abken H. Chimeric Antigen Receptor (CAR) T Cells Releasing Soluble SLAMF6 Isoform 2 Gain Superior Anti-Cancer Cell Functionality in an Auto-Stimulatory Fashion. Cells. 2025; 14(12):901. https://doi.org/10.3390/cells14120901
Chicago/Turabian StyleHarrer, Dennis Christoph, Tim Schlierkamp-Voosen, Markus Barden, Hong Pan, Maria Xydia, Wolfgang Herr, Jan Dörrie, Niels Schaft, and Hinrich Abken. 2025. "Chimeric Antigen Receptor (CAR) T Cells Releasing Soluble SLAMF6 Isoform 2 Gain Superior Anti-Cancer Cell Functionality in an Auto-Stimulatory Fashion" Cells 14, no. 12: 901. https://doi.org/10.3390/cells14120901
APA StyleHarrer, D. C., Schlierkamp-Voosen, T., Barden, M., Pan, H., Xydia, M., Herr, W., Dörrie, J., Schaft, N., & Abken, H. (2025). Chimeric Antigen Receptor (CAR) T Cells Releasing Soluble SLAMF6 Isoform 2 Gain Superior Anti-Cancer Cell Functionality in an Auto-Stimulatory Fashion. Cells, 14(12), 901. https://doi.org/10.3390/cells14120901