
Ultrasound-Mediated Drug Diffusion, Uptake, and Cytotoxicity in a Glioblastoma 3D Tumour Sphere Model

 ,
,  ,
, 
 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Two-Dimensional Cell Culture
2.3. Three-Dimensional Cell Culture
2.4. Ultrasound Probe Device
2.5. Ultrasound Treatment
2.6. TMZ and DOX Treatment
2.7. Cell Viability Assay
2.8. Cell Viability Assay (3D Cell Culture)
2.9. Live/Dead Cell Staining Using Propidium Iodide (PI)
2.10. DOX Sonoporation Analysis
2.11. Statistical Analysis
3. Results and Discussion
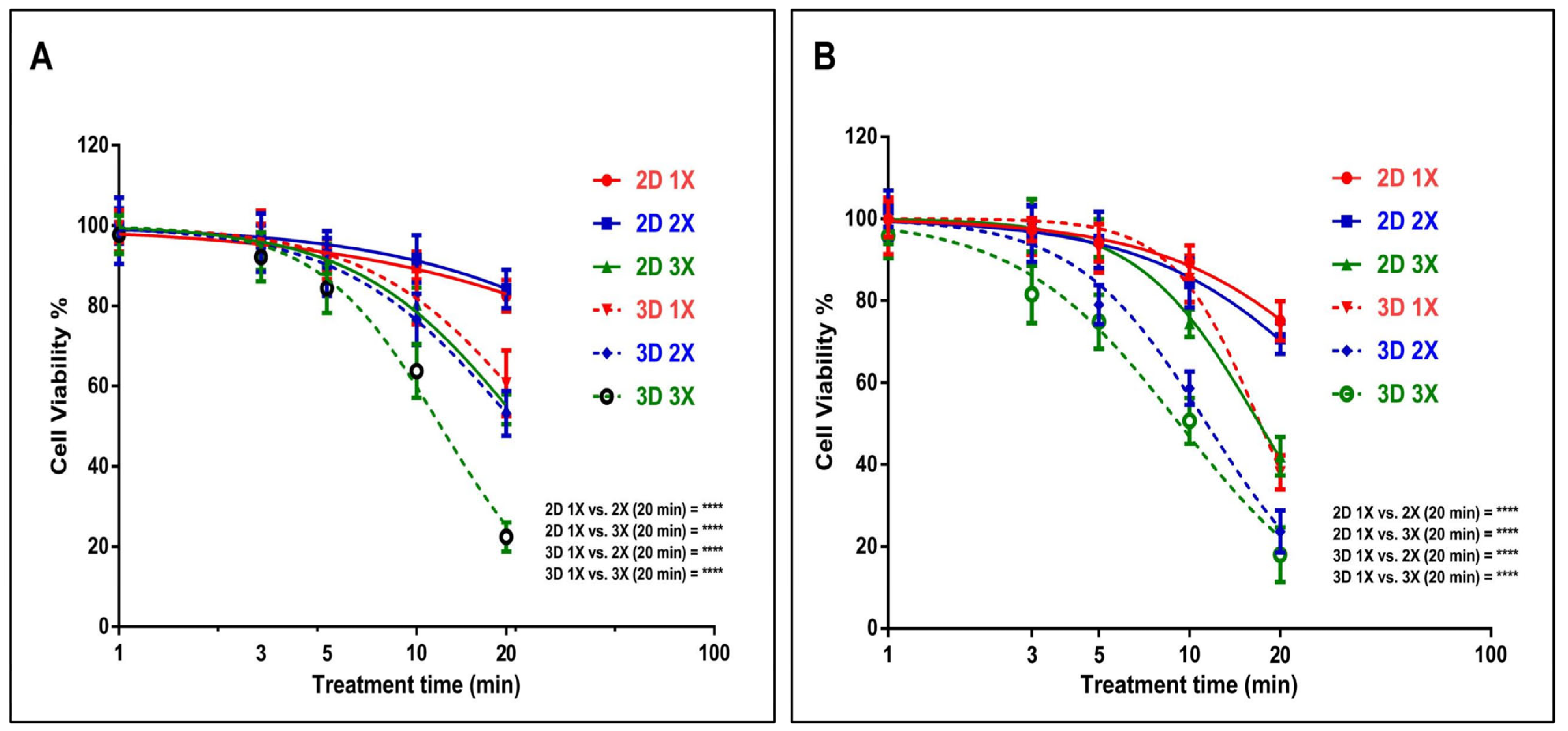
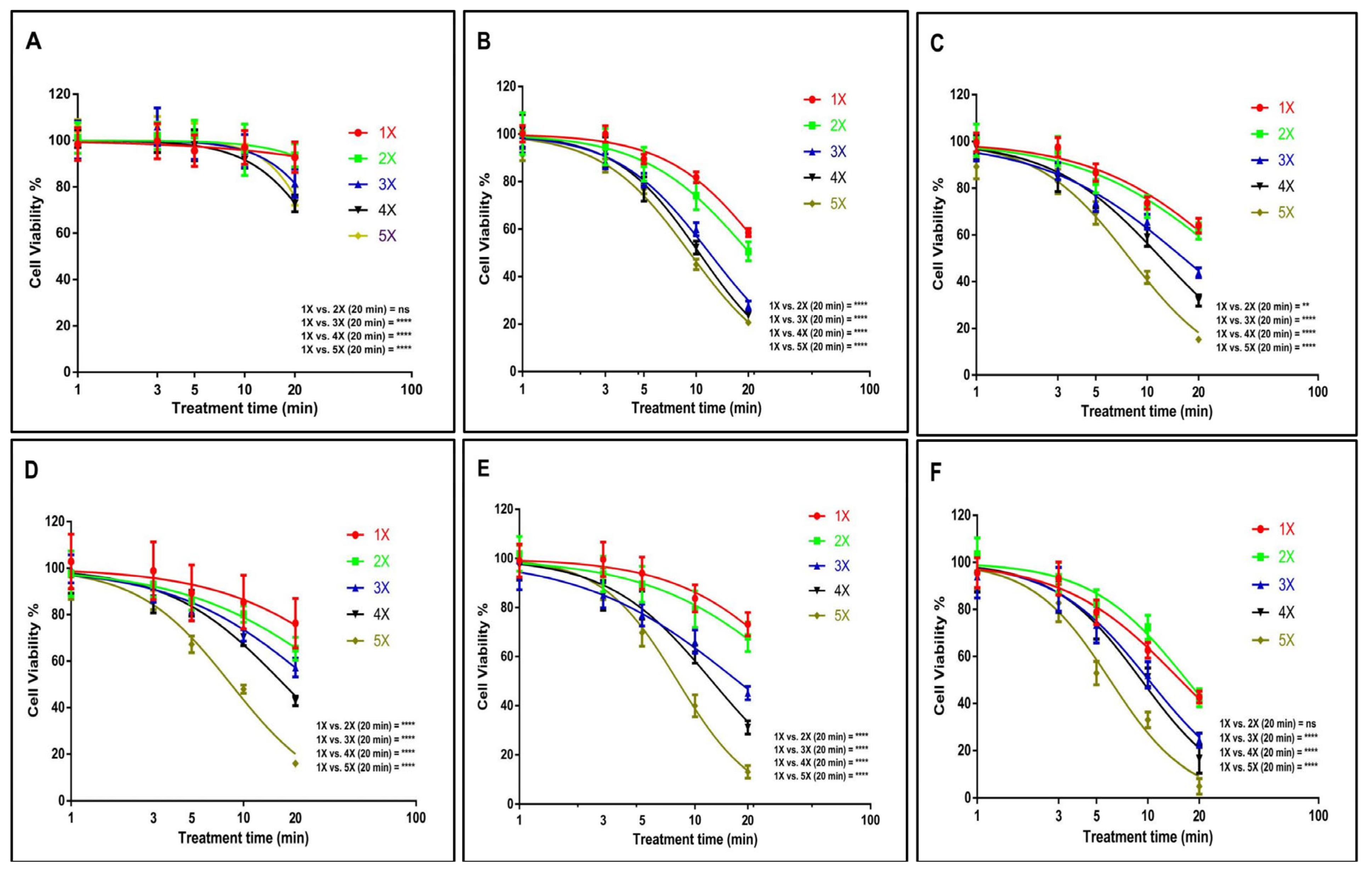
3.1. US Probe Presents Cytotoxicity Towards GBM and EPIDERMOID Carcinoma Cells in a Time- and Dosage-Dependent Manner
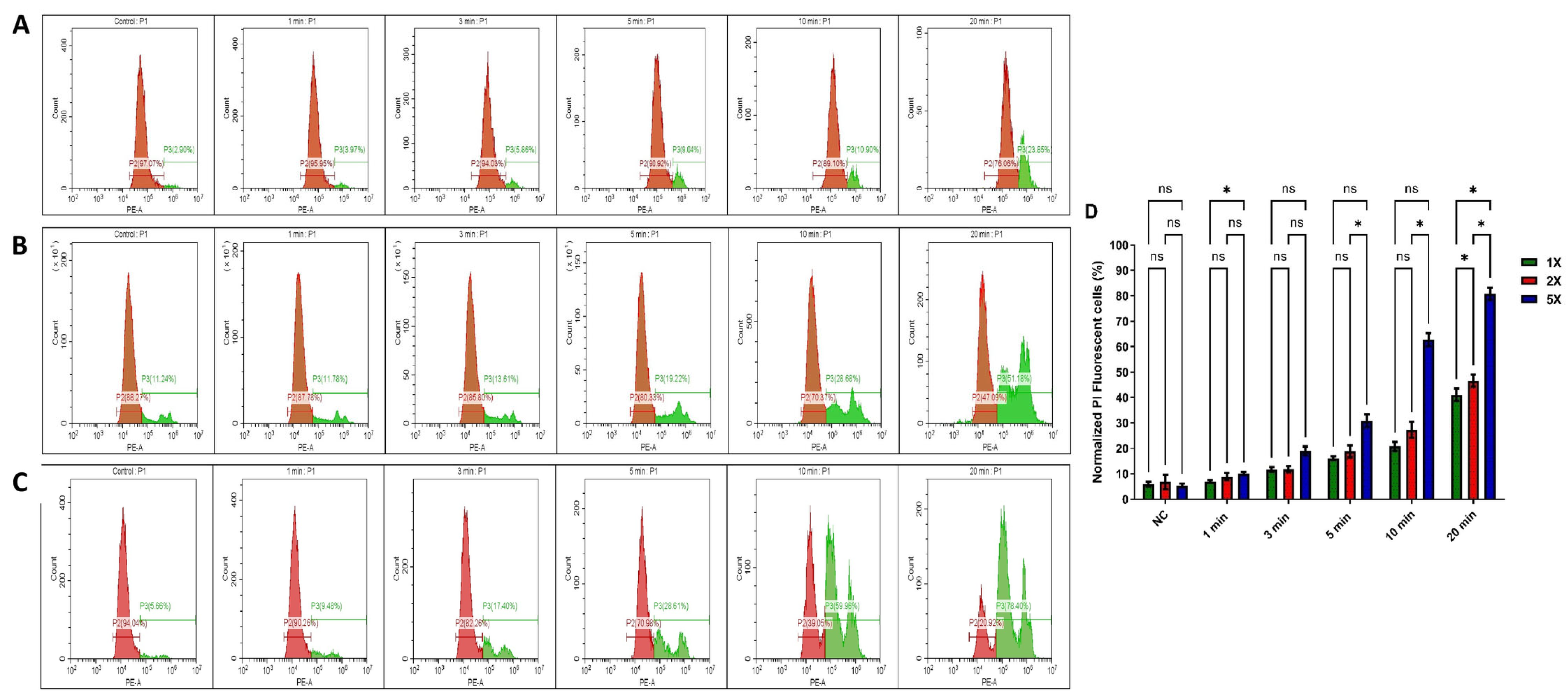
3.2. Impact of US Treatment on Cell Membrane Damage Within Tumour Spheres
3.3. Tumour Sphere Morphological Changes Induced by Ultrasound Probe
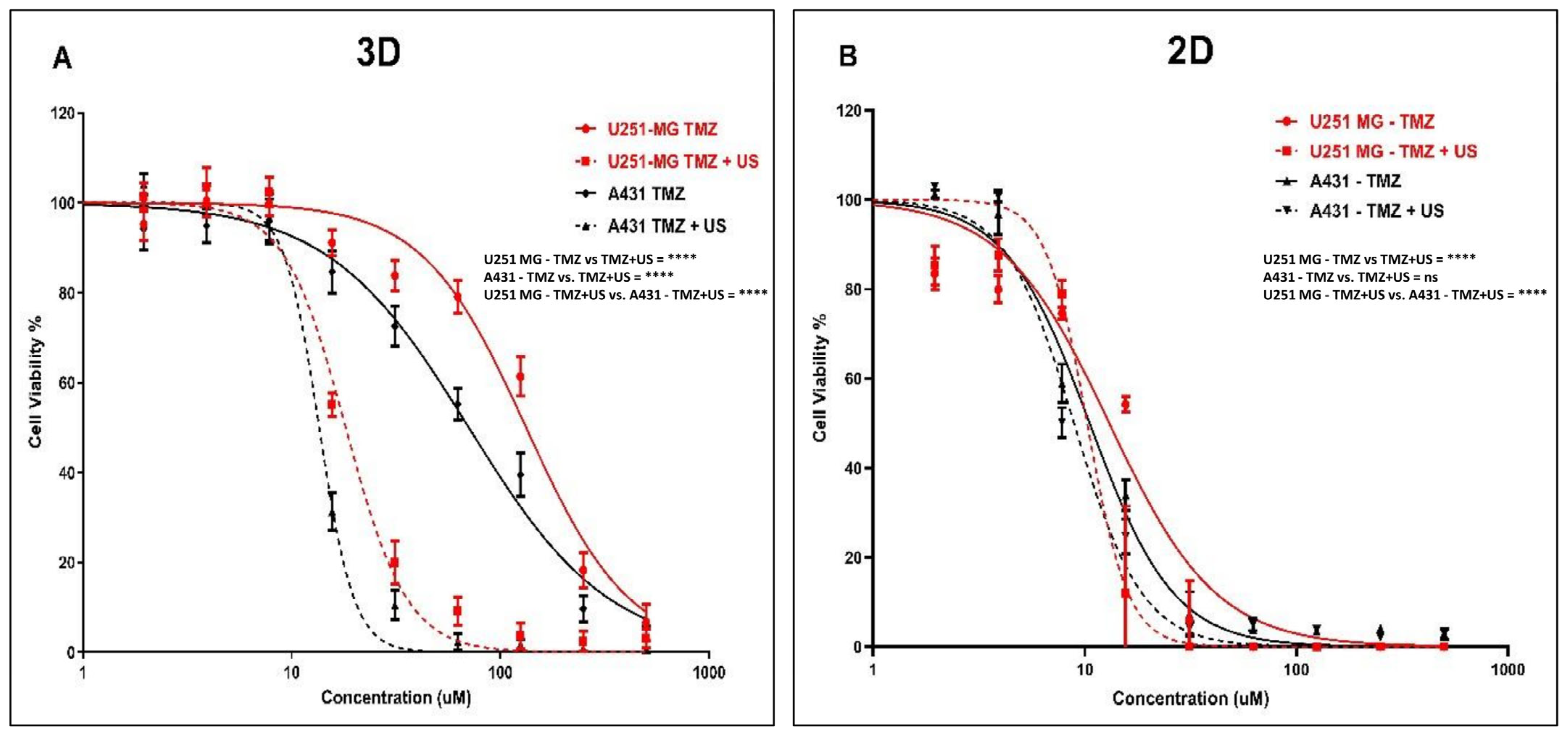
3.4. Effect of US on TMZ Delivery in Human Glioblastoma and Epidermoid Carcinoma Cell Models
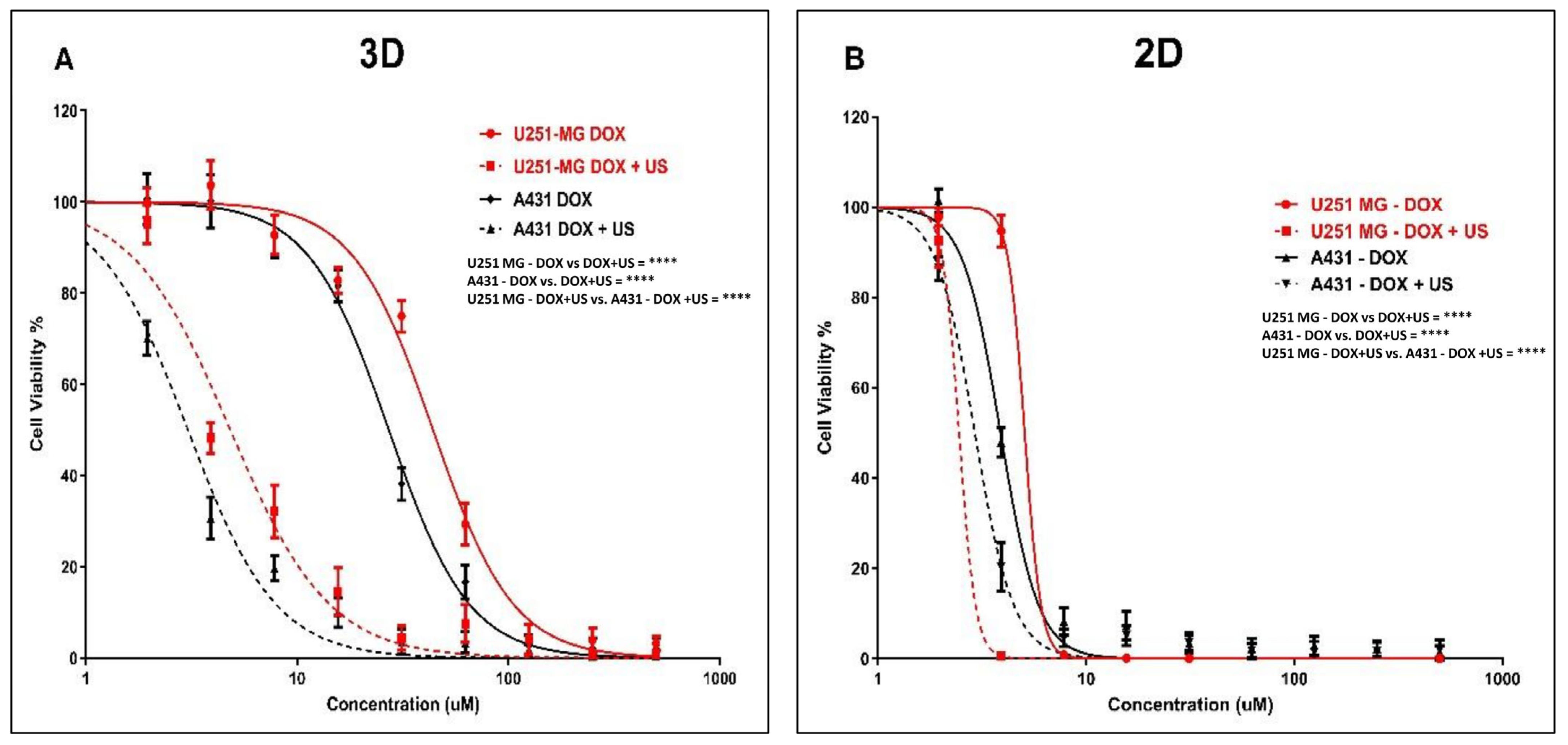
3.5. Effect of US on DOX Delivery in Human Glioblastoma and Epidermoid Carcinoma Cell Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Kumar, N.; Privat-maldonado, A.; Smits, E.; Bogaerts, A. Cold Atmospheric Plasma Increases Temozolomide Sensitivity of Three-dimensional Glioblastoma Spheroids via Oxidative Stress-mediated Dna Damage. Cancers 2021, 13, 1780. [Google Scholar] [CrossRef] [PubMed]
- Wanigasekara, J.; Cullen, P.J.; Bourke, P.; Tiwari, B.; Curtin, J.F. Advances in 3D Culture Systems for Therapeutic Discovery and Development in Brain Cancer. Drug Discov. Today 2023, 28, 103426. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, H.; Wang, P.; Zhang, K.; Geng, X.; Liu, Q.; Wang, X. Tumor Targeting DVDMS-Nanoliposomes for an Enhanced Sonodynamic Therapy of Gliomas. Biomater. Sci. 2019, 7, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, M.; Li, Y.; Sun, Y.; Teng, Y.; Wang, Y.; Duan, Y. The Synergic Antitumor Effects of Paclitaxel and Temozolomide Co-Loaded in MPEG-PLGA Nanoparticles on Glioblastoma Cells. Oncotarget 2016, 7, 20890–20901. [Google Scholar] [CrossRef]
- Wanigasekara, J.; Witharana, C. Applications of Nanotechnology in Drug Delivery and Design—An Insight. Curr. Trends Biotechnol. Pharm. 2016, 10, 78–91. [Google Scholar]
- Aparicio-Blanco, J.; Sanz-Arriazu, L.; Lorenzoni, R.; Blanco-Prieto, M.J. Glioblastoma Chemotherapeutic Agents Used in the Clinical Setting and in Clinical Trials: Nanomedicine Approaches to Improve Their Efficacy. Int. J. Pharm. 2020, 581, 119283. [Google Scholar] [CrossRef]
- Lee, S.; Jeon, H.; Shim, S.; Im, M.; Kim, J.; Kim, J.H.; Lee, B.C. Preclinical Study to Improve Microbubble-Mediated Drug Delivery in Cancer Using an Ultrasonic Probe with an Interchangeable Acoustic Lens. Sci. Rep. 2021, 11, 12654. [Google Scholar] [CrossRef]
- Paškevičiūtė, M.; Januškevičienė, I.; Sakalauskienė, K.; Raišutis, R.; Petrikaitė, V. Evaluation of Low-Intensity Pulsed Ultrasound on Doxorubicin Delivery in 2D and 3D Cancer Cell Cultures. Sci. Rep. 2020, 10, 16161. [Google Scholar] [CrossRef]
- Jimenez-Alcázár, M.; Curiel-Gárcía, A.; Nogales, P.; Perales-Páton, J.; Schuhmacher, A.J.; Galán-Ganga, M.; Zhu, L.; Lowe, S.W.; Al-Shahrour, F.; Squatrito, M. Dianhydrogalactitol Overcomes Multiple Temozolomide Resistance Mechanisms in Glioblastoma. Mol. Cancer Ther. 2021, 20, 1029–1038. [Google Scholar] [CrossRef]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr. Neuropharmacol. 2020, 19, 513–537. [Google Scholar] [CrossRef]
- Fukushima, T.; Takeshima, H.; Kataoka, H. Anti-Glioma Therapy with Temozolomide and Status of the DNA-Repair Gene MGMT. Anticancer Res. 2009, 29, 4845–4854. [Google Scholar] [PubMed]
- Huang, W.; Peng, Y.; Kang, L. Advancements of Non-Invasive Imaging Technologies for the Diagnosis and Staging of Liver Fibrosis: Present and Future. VIEW 2024, 5, 20240010. [Google Scholar] [CrossRef]
- Jagannathan, J.; Sanghvi, N.T.; Crum, L.A.; Yen, C.P.; Medel, R.; Dumont, A.S.; Sheehan, J.P.; Steiner, L.; Jolesz, F.; Kassell, N.F. High-Intensity Focused Ultrasound Surgery of the Brain: Part 1-A Historical Perspective with Modern Applications. Neurosurgery 2009, 64, 201–210. [Google Scholar] [CrossRef]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-Invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood-Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef]
- Wanigasekara, J.; de Carvalho, A.M.A.; Cullen, P.J.; Tiwari, B.; Curtin, J.F. Converging Technologies: Targeting the Hallmarks of Cancer Using Ultrasound and Microbubbles. Trends Cancer 2021, 7, 886–890. [Google Scholar] [CrossRef]
- Lee, S.; Al-Kaabi, L.; Mawart, A.; Khandoker, A.; Alsafar, H.; Jelinek, H.F.; Khalaf, K.; Park, J.H.; Kim, Y.C. Ultrasound-Mediated Drug Delivery by Gas Bubbles Generated from a Chemical Reaction. J. Drug Target. 2018, 26, 172–181. [Google Scholar] [CrossRef]
- Pi, Z.; Huang, Y.; Shen, Y.; Zeng, X.; Hu, Y.; Chen, T.; Li, C.; Yu, H.; Chen, S.; Chen, X. Sonodynamic Therapy on Intracranial Glioblastoma Xenografts Using Sinoporphyrin Sodium Delivered by Ultrasound with Microbubbles. Ann. Biomed. Eng. 2019, 47, 549–562. [Google Scholar] [CrossRef]
- Zarnitsyn, V.; Kamaev, P.; Prausnitz, M. Ultrasound-Enhanced Chemotherapy and Gene Delivery for Glioma Cells. Technol. Cancer Res. Treat. 2007, 6, 433–442. [Google Scholar] [CrossRef]
- Aguiar de Carvalho, A.M.; Scally, L.; Tiwari, B.; Cullen, P.J.; Curtin, J.F. Synergistic Cytotoxicity from Cold Atmospheric Plasma and Ultrasound in Glioma Cells. Plasma Process. Polym. 2022, 19, 2200042. [Google Scholar] [CrossRef]
- Baghbani, F.; Chegeni, M.; Moztarzadeh, F.; Mohandesi, J.A.; Mokhtari-Dizaji, M. Ultrasonic Nanotherapy of Breast Cancer Using Novel Ultrasound-Responsive Alginate-Shelled Perfluorohexane Nanodroplets: In Vitro and in Vivo Evaluation. Mater. Sci. Eng. C 2017, 77, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.K.W.; Sehgal, C.M. A Review of Low-Intensity Ultrasound for Cancer Therapy. Ultrasound Med. Biol. 2015, 41, 905–928. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Zhang, J.; Yuan, C.; Zhang, H.; Hou, X.; Zhang, D. Enhanced ShRNA Delivery by the Combination of Polyethylenimine, Ultrasound, and Nanobubbles in Liver Cancer. Technol. Health Care 2019, 27, S263–S272. [Google Scholar] [CrossRef]
- Kinoshita, M.; Mcdannold, N.; Jolesz, F.A.; Hynynen, K. Noninvasive Localized Delivery of Herceptin to the Mouse Brain by MRI-Guided Focused Ultrasound-Induced Blood-Brain Barrier Disruption. Proc. Natl. Acad. Sci. USA 2006, 103, 11719–11723. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Di Benedetto, A.; Howard, C.M.; Kelly, S.; Nande, R.; Dementieva, Y.; Miranda, M.; Brunetti, A.; Salvatore, M.; Claudio, L.; et al. Eradication of Therapy-Resistant Human Prostate Tumors Using an Ultrasound-Guided Site-Specific Cancer Terminator Virus Delivery Approach. Mol. Ther. 2010, 18, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Amin, M.A.; Campian, J.L. Glioblastoma Stem Cells at the Nexus of Tumor Heterogeneity, Immune Evasion, and Therapeutic Resistance. Cells 2025, 14, 562. [Google Scholar] [CrossRef]
- Nayak, P.; Bentivoglio, V.; Varani, M.; Signore, A. Three-Dimensional In Vitro Tumor Spheroid Models for Evaluation of Anticancer Therapy: Recent Updates. Cancers 2023, 15, 4846. [Google Scholar] [CrossRef]
- Momoli, C.; Costa, B.; Lenti, L.; Tubertini, M.; Parenti, M.; Martella, E.; Varchi, G.; Ferroni, C. The Evolution of Anticancer 3D In Vitro Models: The Potential Role of Machine Learning and AI in the Next Generation of Animal-Free Experiments. Cancers 2025, 17, 700. [Google Scholar] [CrossRef]
- Dave, R.; Pandey, K.; Patel, R.; Gour, N.; Bhatia, D. Leveraging 3D Cell Culture and AI Technologies for Next-Generation Drug Discovery. Cell Biomater. 2025, 1, 100050. [Google Scholar] [CrossRef]
- Cha, Y.S.; Michaels, A.; Wang, J.Z.; Niu, Y.; Lin, Y.; Zhu, L.; Zhu, X.; Wang, K.; Murray, M.; Zhou, F. Recent Advances in 3D Cell Culture Models in Cancer Drug Development. J. Pharm. Investig. 2025, 1–17. [Google Scholar] [CrossRef]
- Wanigasekara, J.; Barcia, C.; Cullen, P.J.; Tiwari, B.; Curtin, J.F. Plasma Induced Reactive Oxygen Species-Dependent Cytotoxicity in Glioblastoma 3D Tumourspheres. Plasma Process. Polym. 2022, 19, 2100157. [Google Scholar] [CrossRef]
- Carroll, J.C.; Tiwari, B.; Curtin, J.F.; Wanigasekara, J. U-251MG Spheroid Generation Using Low Attachment Plate Method Protocol. Available online: https://www.protocols.io/view/u-251mg-spheroid-generation-using-low-attachment-p-bszmnf46 (accessed on 1 June 2025).
- Wanigasekara, J.; Carroll, L.J.; Cullen, P.J.; Tiwari, B.; Curtin, J.F. Three-Dimensional (3D) In Vitro Cell Culture Protocols to Enhance Glioblastoma Research. PLoS ONE 2023, 18, e0276248. [Google Scholar] [CrossRef]
- Field, A.; Tiwari, B.; Curtin, J.; Mondala, J.; Wanigasekara, J. Ultrasound 96 Probe Device Protocol for Cancer Cell Treatment; Protocols.io: Berkeley, CA, USA, 2023. [Google Scholar] [CrossRef]
- Chhetri, A.; Rispoli, J.V.; Lelièvre, S.A. 3D Cell Culture for the Study of Microenvironment-Mediated Mechanostimuli to the Cell Nucleus: An Important Step for Cancer Research. Front. Mol. Biosci. 2021, 8, 628386. [Google Scholar] [CrossRef] [PubMed]
- Foglietta, F.; Canaparo, R.; Muccioli, G.; Terreno, E.; Serpe, L. Methodological Aspects and Pharmacological Applications of Three-Dimensional Cancer Cell Cultures and Organoids. Life Sci. 2020, 254, 117784. [Google Scholar] [CrossRef]
- Reidy, E.; Leonard, N.A.; Treacy, O.; Ryan, A.E. Cancers A 3D View of Colorectal Cancer Models in Predicting Therapeutic Responses and Resistance. Cancers 2021, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Martinez-Hackert, E. Determination of Half-Maximal Inhibitory Concentration Using Biosensor-Based Protein Interaction Analysis. Anal. Biochem. 2016, 508, 97–103. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning from 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Landgraf, L.; Kozlowski, A.; Zhang, X.; Fournelle, M.; Becker, F.J.; Tretbar, S.; Melzer, A. Focused Ultrasound Treatment of a Spheroid In Vitro Tumour Model. Cells 2022, 11, 1518. [Google Scholar] [CrossRef]
- Rwei, P.; Alex Gong, C.S.; Luo, L.J.; Lin, M.B.; Lai, J.Y.; Liu, H.L. In Vitro Investigation of Ultrasound-Induced Oxidative Stress on Human Lens Epithelial Cells. Biochem. Biophys. Res. Commun. 2017, 482, 954–960. [Google Scholar] [CrossRef]
- Pefani-Antimisiari, K.; Athanasopoulos, D.K.; Marazioti, A.; Sklias, K.; Rodi, M.; de Lastic, A.L.; Mouzaki, A.; Svarnas, P.; Antimisiaris, S.G. Synergistic Effect of Cold Atmospheric Pressure Plasma and Free or Liposomal Doxorubicin on Melanoma Cells. Sci. Rep. 2021, 11, 14788. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Lee, Y.-S.; Kim, D.-K. Doxorubicin Exerts Cytotoxic Effects through Cell Cycle Arrest and Fas-Mediated Cell Death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Wanigasekara, J.; Cullen, P.J.; Tiwari, B.; Curtin, J.F. Synergistic Cytotoxicity Effect of Ultrasound and Plasma Microbubble in Glioblastoma 3D Tumor Sphere Model. Plasma Process. Polym. 2024, 21, 2300185. [Google Scholar] [CrossRef]
- Sampson, A.; Peterson, B.G.; Tan, K.W.; Iram, S.H. Doxorubicin as a Fluorescent Reporter Identifies Novel MRP1 (ABCC1) Inhibitors Missed by Calcein-Based High Content Screening of Anticancer Agents. Biomed. Pharmacother. 2019, 118, 109289. [Google Scholar] [CrossRef]
- van Rijt, A.; Stefanek, E.; Valente, K. Preclinical Testing Techniques: Paving the Way for New Oncology Screening Approaches. Cancers 2023, 15, 4466. [Google Scholar] [CrossRef]
- Qian, L.; Lu, X.; Haris, P.; Zhu, J.; Li, S.; Yang, Y. Enhancing Clinical Trial Outcome Prediction with Artificial Intelligence: A Systematic Review. Drug Discov. Today 2025, 30, 104332. [Google Scholar] [CrossRef]
- White, J.; White, M.P.J.; Wickremesekera, A.; Peng, L.; Gray, C. The Tumour Microenvironment, Treatment Resistance and Recurrence in Glioblastoma. J. Transl. Med. 2024, 22, 540. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanigasekara, J.; Mondala, J.R.M.; Cullen, P.J.; Tiwari, B.K.; Kinsella, G.K.; Curtin, J.F. Ultrasound-Mediated Drug Diffusion, Uptake, and Cytotoxicity in a Glioblastoma 3D Tumour Sphere Model. Cells 2025, 14, 886. https://doi.org/10.3390/cells14120886
Wanigasekara J, Mondala JRM, Cullen PJ, Tiwari BK, Kinsella GK, Curtin JF. Ultrasound-Mediated Drug Diffusion, Uptake, and Cytotoxicity in a Glioblastoma 3D Tumour Sphere Model. Cells. 2025; 14(12):886. https://doi.org/10.3390/cells14120886
Chicago/Turabian StyleWanigasekara, Janith, Julie Rose Mae Mondala, Patrick J. Cullen, Brijesh K. Tiwari, Gemma K. Kinsella, and James F. Curtin. 2025. "Ultrasound-Mediated Drug Diffusion, Uptake, and Cytotoxicity in a Glioblastoma 3D Tumour Sphere Model" Cells 14, no. 12: 886. https://doi.org/10.3390/cells14120886
APA StyleWanigasekara, J., Mondala, J. R. M., Cullen, P. J., Tiwari, B. K., Kinsella, G. K., & Curtin, J. F. (2025). Ultrasound-Mediated Drug Diffusion, Uptake, and Cytotoxicity in a Glioblastoma 3D Tumour Sphere Model. Cells, 14(12), 886. https://doi.org/10.3390/cells14120886