
5′ DREDGE: Direct Repeat-Enabled Downregulation of Gene Expression via the 5′ UTR of Target Genes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning of Expression Constructs
2.1.1. Vectors for Transient Transfection Experiments
2.1.2. Vectors for Dox-Regulatable Co-Expression of Cas RNases, mCherry, and Neor
2.2. Cell Culture
2.3. FACS and Flow Cytometry
2.4. Real-Time PCR
2.5. Insertion of a Csy4 DR into the 5′ UTR of the CTSD Gene
2.6. CatD Activity Assays
3. Results
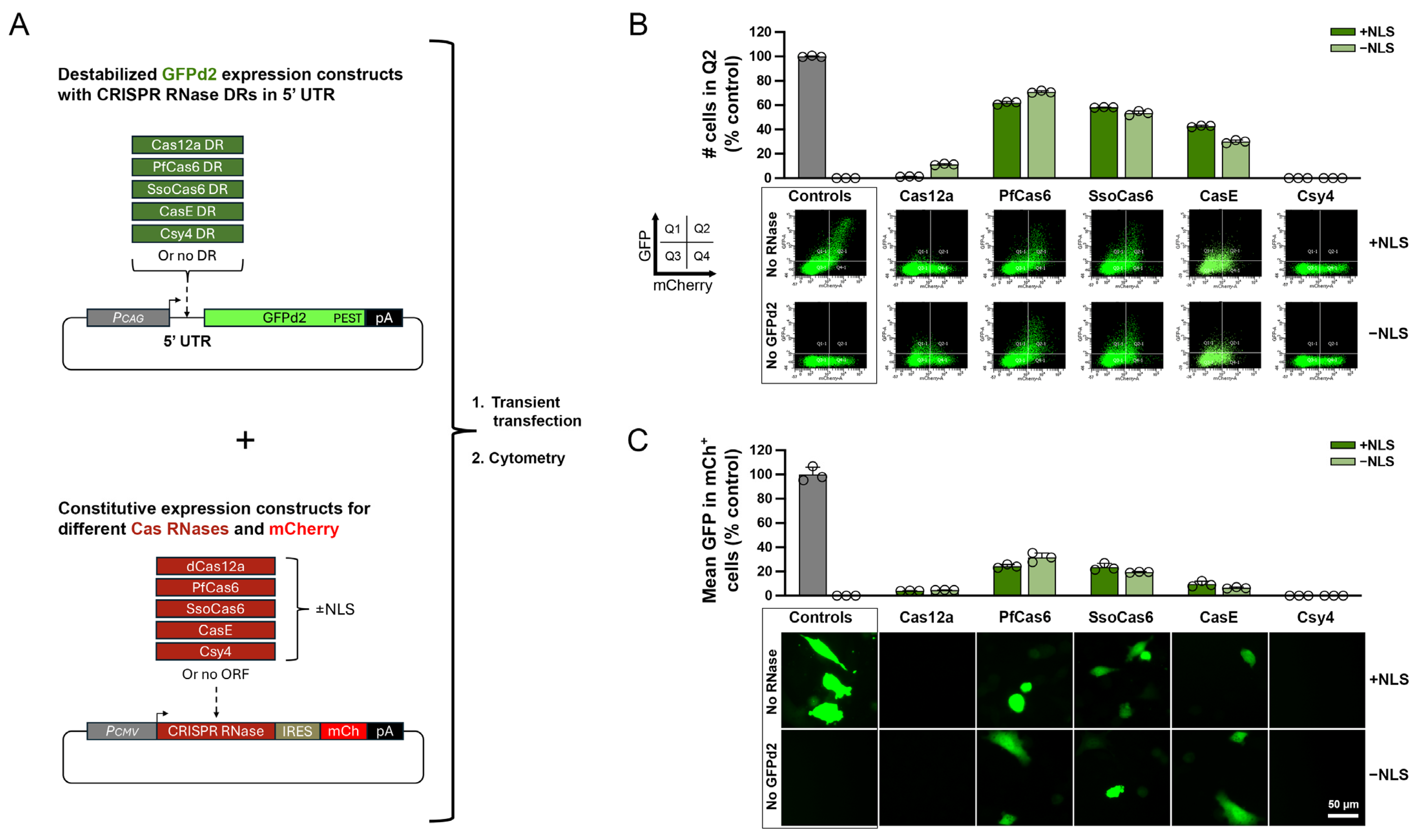
3.1. Selection and Screening of Candidate Cas RNases for 5′ DREDGE
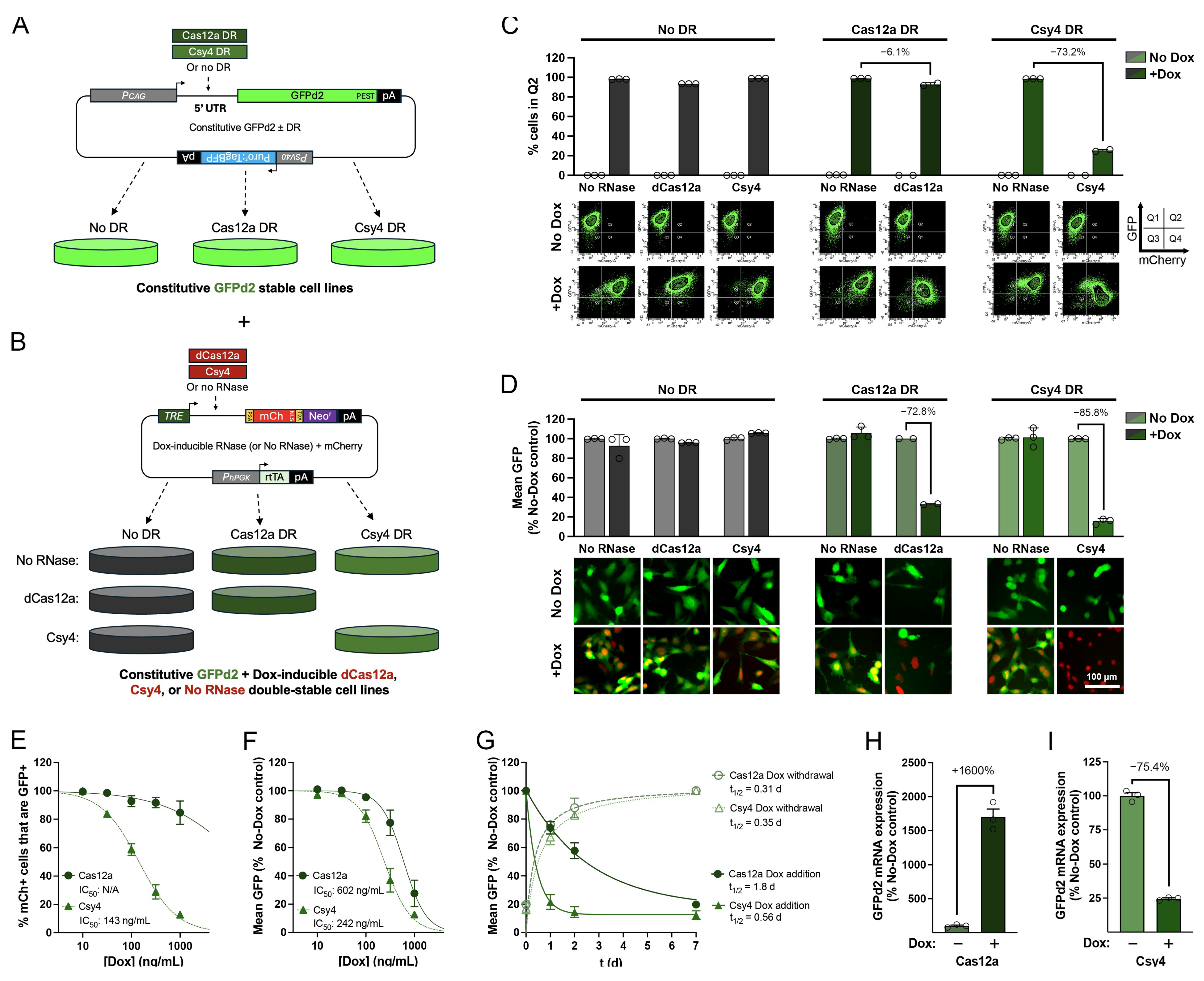
3.2. Dox-Regulatable Gene Expression by 5′ DREDGE Using dCas12a and Csy4
3.3. Downregulation of an Endogenous Gene by 5′ DREDGE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charpentier, E.; Richter, H.; van der Oost, J.; White, M.F. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol. Rev. 2015, 39, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M.L.; Doudna, J.A. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem. Sci. 2015, 40, 58–66. [Google Scholar] [CrossRef]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.J.; Terron, H.M.; Burgard, L.A.; Maranan, D.S.; Butler, D.D.; Wiseman, A.; LaFerla, F.M.; Lane, S.; Leissring, M.A. Targeted control of gene expression using CRISPR-associated endoribonucleases. Cells 2025, 14, 543. [Google Scholar] [CrossRef]
- Chen, C.Y.; Shyu, A.B. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip. Rev. RNA 2011, 2, 167–183. [Google Scholar] [CrossRef]
- Guo, L.Y.; Bian, J.; Davis, A.E.; Liu, P.; Kempton, H.R.; Zhang, X.; Chemparathy, A.; Gu, B.; Lin, X.; Rane, D.A.; et al. Multiplexed genome regulation in vivo with hyper-efficient Cas12a. Nat. Cell Biol. 2022, 24, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Avila-Bonilla, R.G.; Macias, S. The molecular language of RNA 5′ ends: Guardians of RNA identity and immunity. RNA 2024, 30, 327–336. [Google Scholar] [CrossRef]
- Borden, K.L.B.; Volpon, L. The diversity, plasticity, and adaptability of cap-dependent translation initiation and the associated machinery. RNA Biol. 2020, 17, 1239–1251. [Google Scholar] [CrossRef]
- Mars, J.C.; Culjkovic-Kraljacic, B.; Borden, K.L.B. eIF4E orchestrates mRNA processing, RNA export and translation to modify specific protein production. Nucleus 2024, 15, 2360196. [Google Scholar] [CrossRef]
- He, F.; Jacobson, A. Eukaryotic mRNA decapping factors: Molecular mechanisms and activity. FEBS J. 2023, 290, 5057–5085. [Google Scholar] [CrossRef]
- Kramer, S.; McLennan, A.G. The complex enzymology of mRNA decapping: Enzymes of four classes cleave pyrophosphate bonds. Wiley Interdiscip. Rev. RNA 2019, 10, e1511. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Haurwitz, R.E.; Doudna, J.A. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA 2012, 18, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Cepko, C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 2007, 104, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef]
- Suire, C.N.; Abdul-Hay, S.O.; Sahara, T.; Kang, D.; Brizuela, M.K.; Saftig, P.; Dickson, D.W.; Rosenberry, T.L.; Leissring, M.A. Cathepsin D regulates cerebral Abeta42/40 ratios via differential degradation of Abeta42 and Abeta40. Alzheimer’s Res. Ther. 2020, 12, 80. [Google Scholar] [CrossRef]
- Terron, H.M.; Maranan, D.S.; Burgard, L.A.; LaFerla, F.M.; Lane, S.; Leissring, M.A. A dual-function “TRE-Lox” system for genetic deletion or reversible, titratable, and near-complete downregulation of cathepsin D. Int. J. Mol. Sci. 2023, 24, 6745. [Google Scholar] [CrossRef]
- Magnusson, J.P.; Rios, A.R.; Wu, L.; Qi, L.S. Enhanced Cas12a multi-gene regulation using a CRISPR array separator. Elife 2021, 10, e66406. [Google Scholar] [CrossRef]
- Loew, R.; Heinz, N.; Hampf, M.; Bujard, H.; Gossen, M. Improved Tet-responsive promoters with minimized background expression. BMC Biotechnol. 2010, 10, 81. [Google Scholar] [CrossRef]
- Terron, H.M.; Parikh, S.J.; Abdul-Hay, S.O.; Sahara, T.; Kang, D.; Dickson, D.W.; Saftig, P.; LaFerla, F.M.; Lane, S.; Leissring, M.A. Prominent tauopathy and intracellular beta-amyloid accumulation triggered by genetic deletion of cathepsin D: Implications for Alzheimer disease pathogenesis. Alzheimer’s Res. Ther. 2024, 16, 70. [Google Scholar] [CrossRef]
- Suire, C.N.; Leissring, M.A. Cathepsin D: A candidate link between amyloid beta-protein and tauopathy in Alzheimer disease. J. Exp. Neurol. 2021, 2, 10–15. [Google Scholar] [CrossRef]
- Chen, J.; Lin, X.; Xiang, W.; Chen, Y.; Zhao, Y.; Huang, L.; Liu, L. DNA target binding-induced pre-crRNA processing in type II and V CRISPR-Cas systems. Nucleic Acids Res. 2025, 53, gkae1241. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ren, K.; Qiu, X.; Zheng, J.; Guo, M.; Guan, X.; Liu, H.; Li, N.; Zhang, B.; Yang, D.; et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 2016, 532, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Sinan, S.; Appleby, N.M.; Chou, C.W.; Finkelstein, I.J.; Russell, R. Kinetic dissection of pre-crRNA binding and processing by CRISPR-Cas12a. RNA 2024, 30, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qiu, X.; Zhou, Y.; Luo, Z.; Zhu, L.; Shao, J.; Xie, M.; Wang, H. A trigger-inducible split-Csy4 architecture for programmable RNA modulation. Nucleic Acids Res. 2025, 53, gkae1319. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parikh, S.J.; Terron, H.M.; Burgard, L.A.; Butler, D.D.; LaFerla, F.M.; Lane, S.; Leissring, M.A. 5′ DREDGE: Direct Repeat-Enabled Downregulation of Gene Expression via the 5′ UTR of Target Genes. Cells 2025, 14, 866. https://doi.org/10.3390/cells14120866
Parikh SJ, Terron HM, Burgard LA, Butler DD, LaFerla FM, Lane S, Leissring MA. 5′ DREDGE: Direct Repeat-Enabled Downregulation of Gene Expression via the 5′ UTR of Target Genes. Cells. 2025; 14(12):866. https://doi.org/10.3390/cells14120866
Chicago/Turabian StyleParikh, Sagar J., Heather M. Terron, Luke A. Burgard, Dylan D. Butler, Frank M. LaFerla, Shelley Lane, and Malcolm A. Leissring. 2025. "5′ DREDGE: Direct Repeat-Enabled Downregulation of Gene Expression via the 5′ UTR of Target Genes" Cells 14, no. 12: 866. https://doi.org/10.3390/cells14120866
APA StyleParikh, S. J., Terron, H. M., Burgard, L. A., Butler, D. D., LaFerla, F. M., Lane, S., & Leissring, M. A. (2025). 5′ DREDGE: Direct Repeat-Enabled Downregulation of Gene Expression via the 5′ UTR of Target Genes. Cells, 14(12), 866. https://doi.org/10.3390/cells14120866