
Induced Mitophagy Promotes Cell Cycle Re-Entry in Adult Cardiomyocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Adult CM
2.2. Induction of Mitophagy in Adult CMs via Parkin Transfection and CCCP Treatment
2.3. Immunofluorescence Analysis
2.4. Adult CM Proliferation
2.5. Analysis of Mitochondrial Mass
2.6. Transmission Electron Microscopy
2.7. Analysis of Oxidative Stress
2.8. Statistical Analysis
3. Results
3.1. Mitophagy Regulator Screening Reveals CCCP as a Potent Inducer of Adult Cardiomyocyte Proliferation
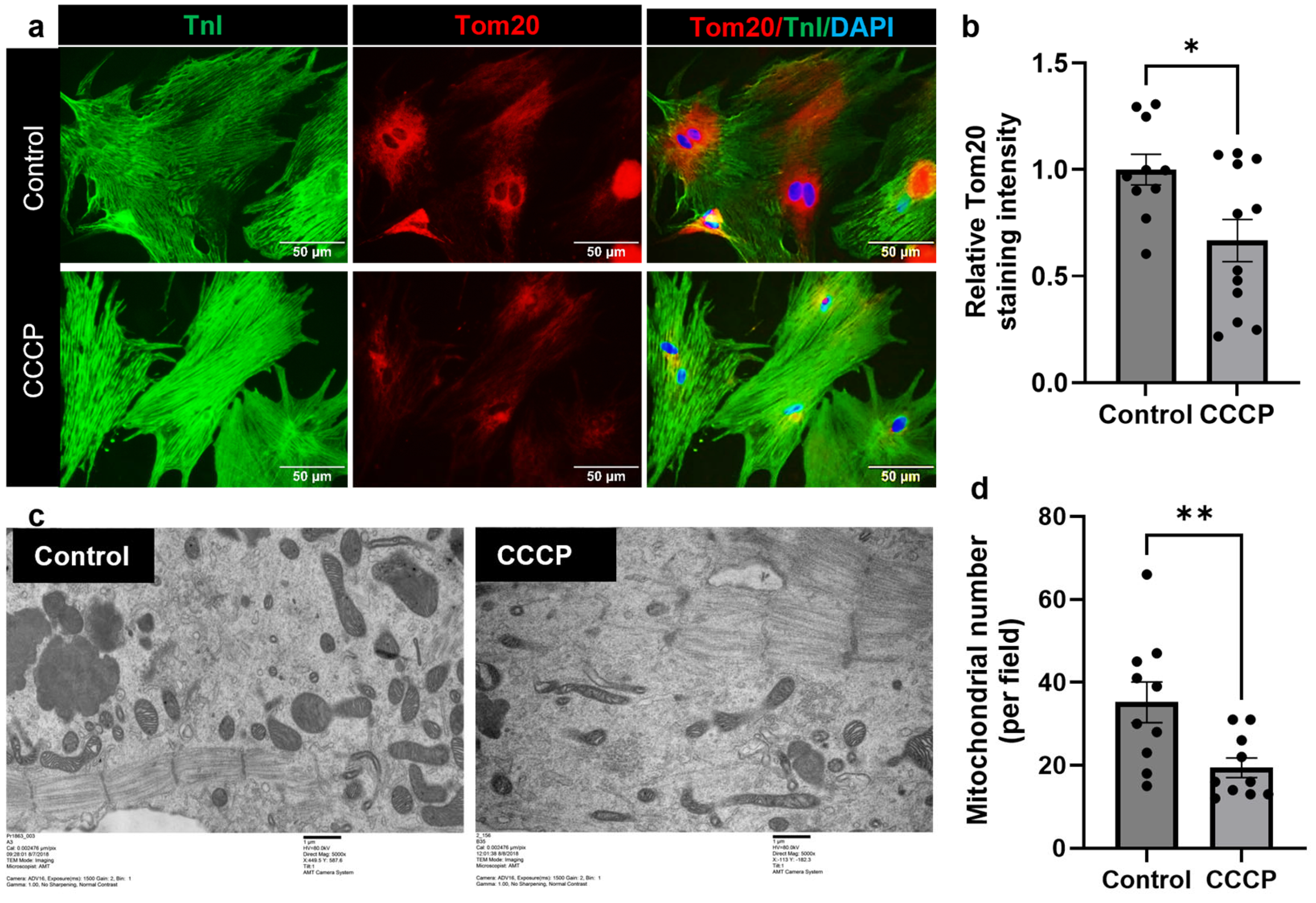
3.2. CCCP Treatment Decreases Mitochondria in Adult Cardiomyocyte

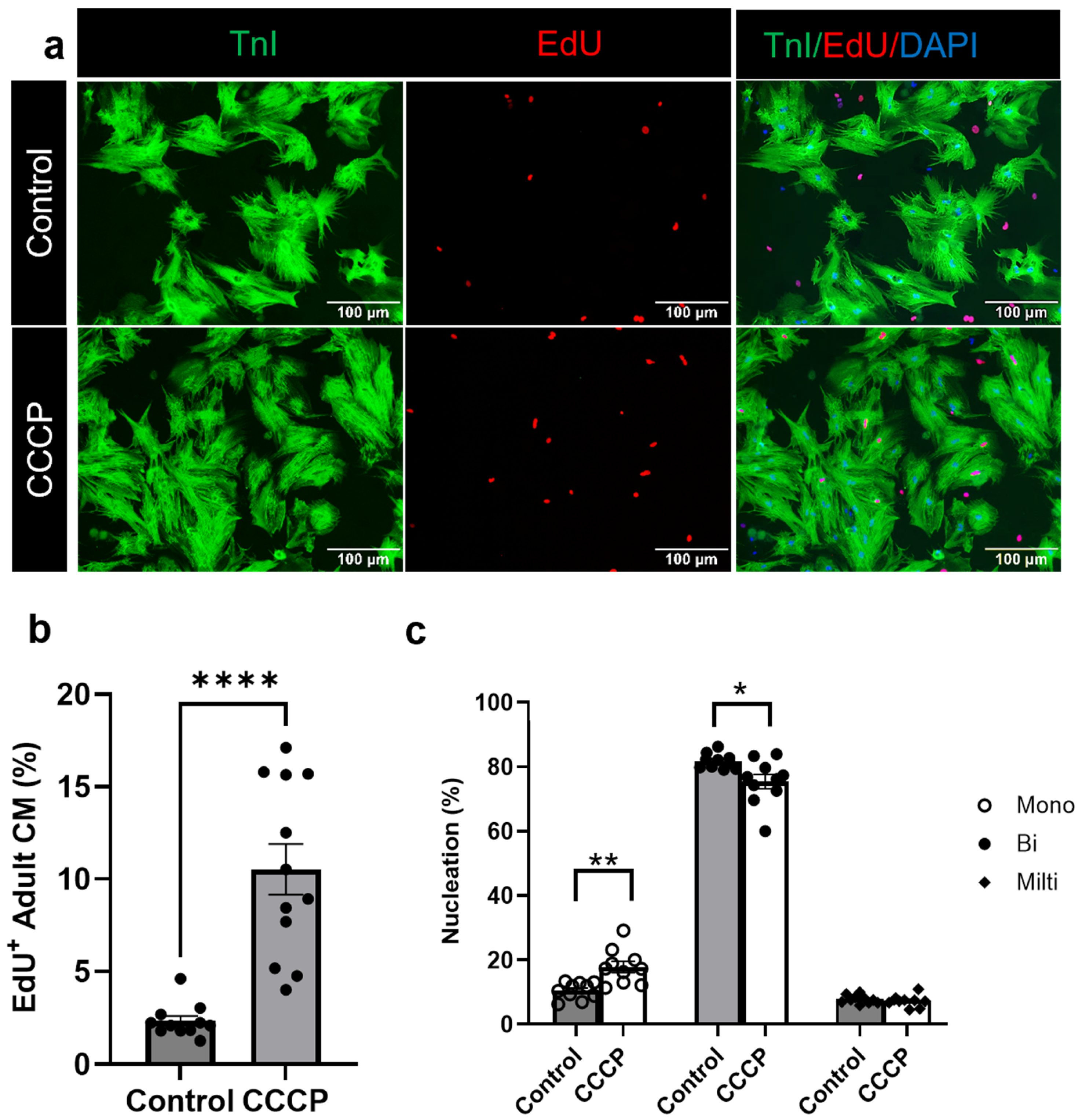
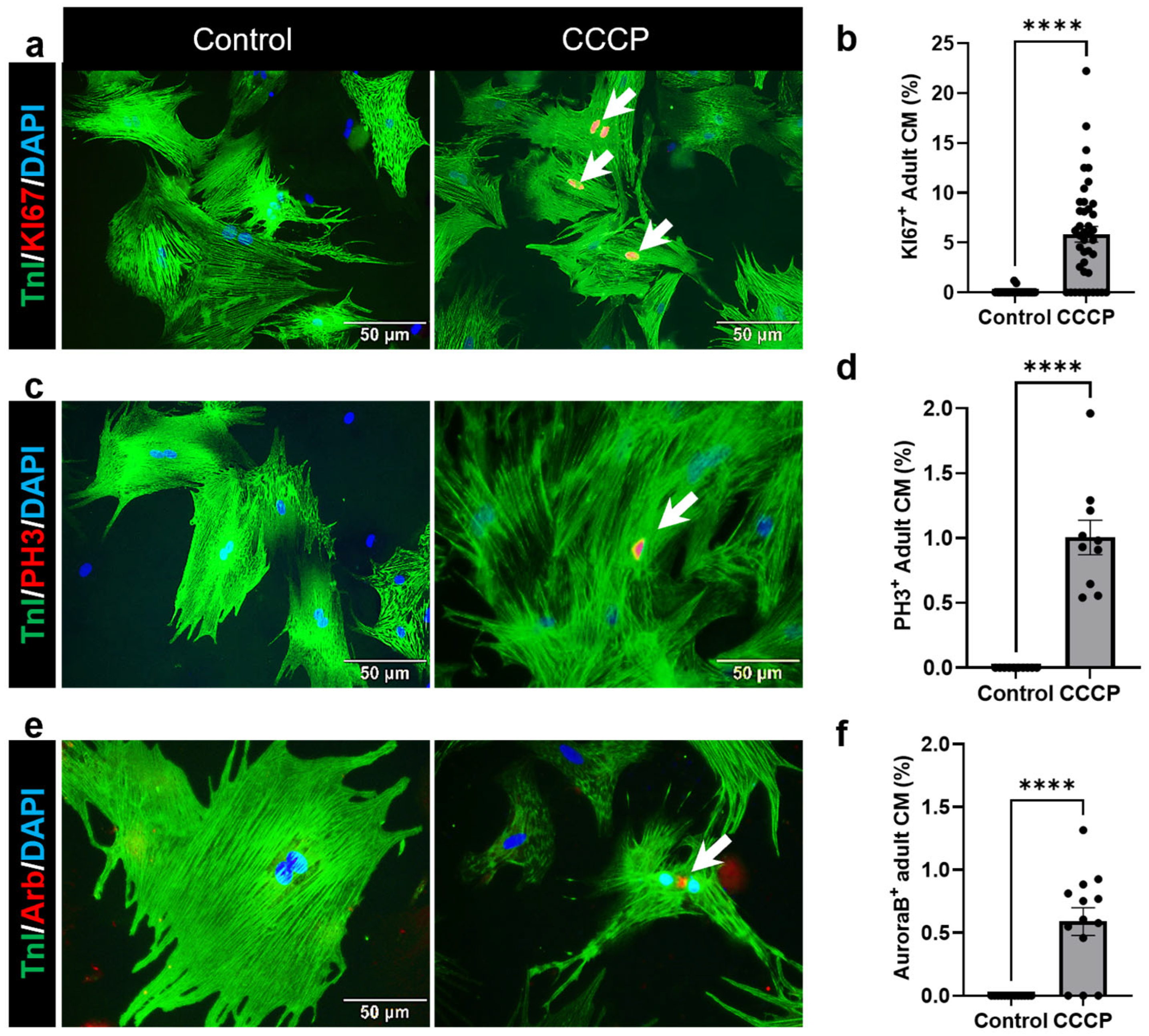
3.3. Induced Mitophagy Promotes Adult Cardiomyocyte Cell Cycle Re-Entry
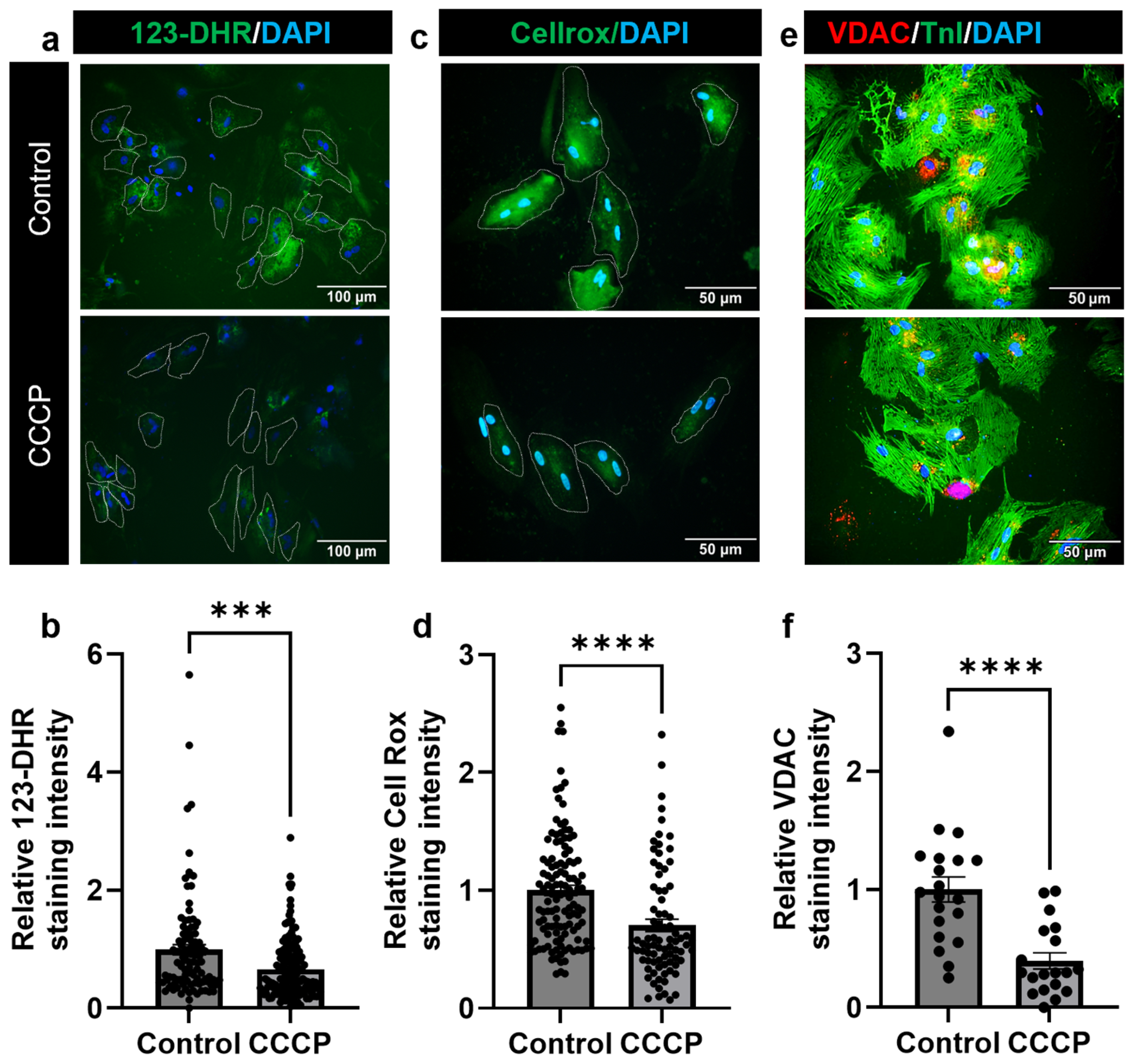
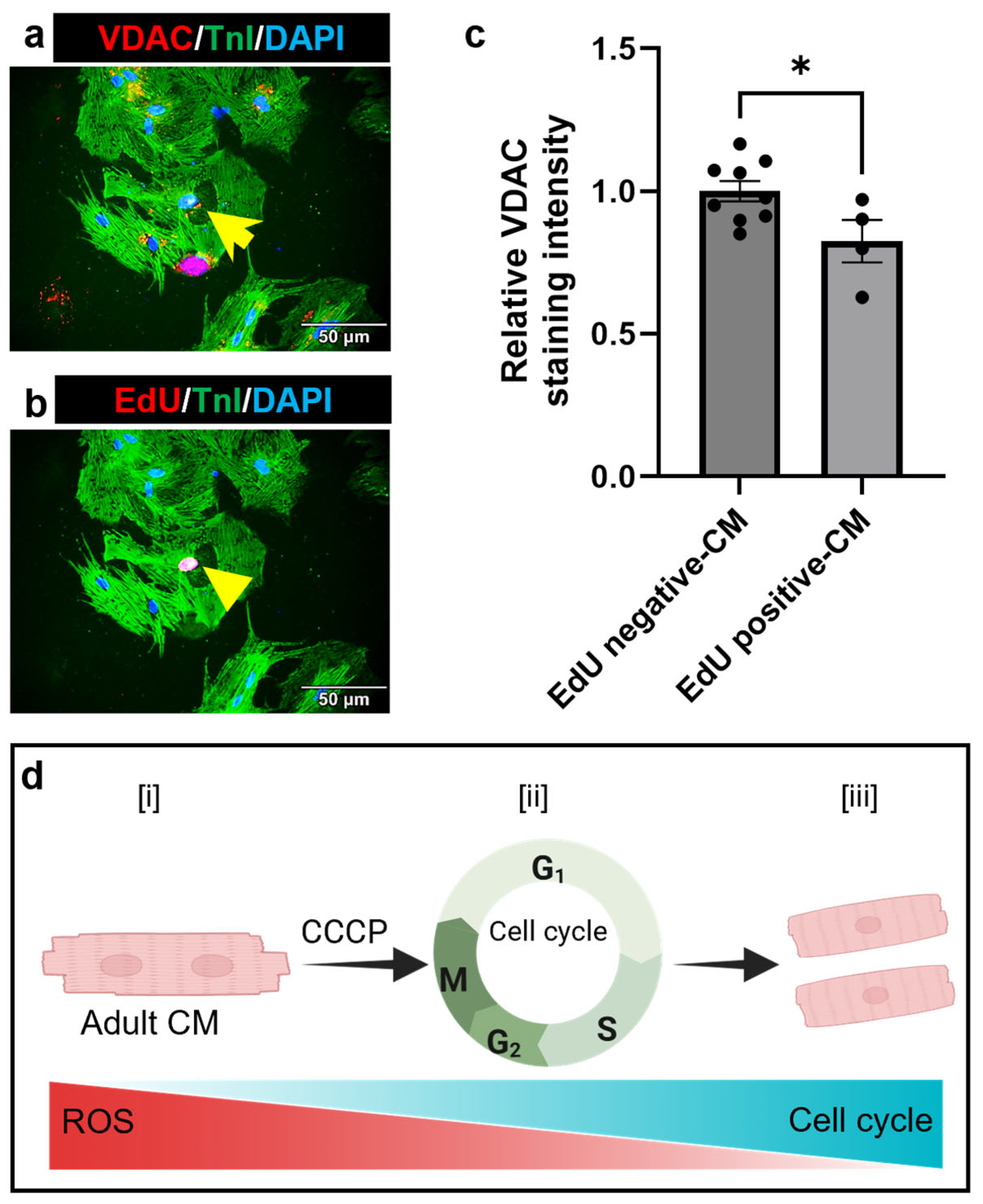
3.4. Parkin and CCCP-Mediated Induced Mitophagy Results in Reduced Oxidative Stress in Adult Cardiomyocyte
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tousoulis, D.; Androulakis, E.; Kontogeorgou, A.; Papageorgiou, N.; Charakida, M.; Siama, K.; Latsios, G.; Siasos, G.; Kampoli, A.M.; Tourikis, P.; et al. Insight to the pathophysiology of stable angina pectoris. Curr. Pharm. Des. 2013, 19, 1593–1600. [Google Scholar] [PubMed]
- Gonzalez-Rosa, J.M.; Martin, V.; Peralta, M.; Torres, M.; Mercader, N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development 2011, 138, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Parente, V.; Balasso, S.; Pompilio, G.; Verduci, L.; Colombo, G.I.; Milano, G.; Guerrini, U.; Squadroni, L.; Cotelli, F.; Pozzoli, O.; et al. Hypoxia/reoxygenation cardiac injury and regeneration in zebrafish adult heart. PLoS ONE 2013, 8, e53748. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Panáková, D.; Kikuchi, K.; Holdway, J.E.; Gemberling, M.; Burris, J.S.; Singh, S.P.; Dickson, A.L.; Lin, Y.F.; Sabeh, M.K.; et al. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 2011, 138, 3421–3430. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003, 5, 741–747. [Google Scholar] [CrossRef]
- Fisher, D.J.; Heymann, M.A.; Rudolph, A.M. Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. Am. J. Physiol. 1980, 238, H399–405. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Gertz, E.W.; Neese, R.A.; Gruenke, L.D.; Morris, D.L.; Craig, J.C. Metabolic fate of extracted glucose in normal human myocardium. J. Clin. Investig. 1985, 76, 1819–1827. [Google Scholar] [CrossRef]
- Gertz, E.W.; Wisneski, J.A.; Stanley, W.C.; Neese, R.A. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Investig. 1988, 82, 2017–2025. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Collins-Nakai, R.L.; Itoi, T. Developmental changes in energy substrate use by the heart. Cardiovasc. Res. 1992, 26, 1172–1180. [Google Scholar] [CrossRef]
- Roesner, A.; Hankeln, T.; Burmester, T. Hypoxia induces a complex response of globin expression in zebrafish (Danio rerio). J. Exp. Biol. 2006, 209 Pt 11, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Rees, B.B.; Sudradjat, F.A.; Love, J.W. Acclimation to hypoxia increases survival time of zebrafish, Danio rerio, during lethal hypoxia. J. Exp. Zool. 2001, 289, 266–272. [Google Scholar] [CrossRef]
- Dawes, G.S.; Mott, J.C.; Widdicombe, J.G. The foetal circulation in the lamb. J. Physiol. 1954, 126, 563–587. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Dismukes, G.C.; Klimov, V.V.; Baranov, S.V.; Kozlov, Y.N.; DasGupta, J.; Tyryshkin, A. The origin of atmospheric oxygen on Earth: The innovation of oxygenic photosynthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 2170–2175. [Google Scholar] [CrossRef]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Chandel, N.S.; Jasper, H.; Ho, T.T.; Passegué, E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol. 2016, 18, 823–832. [Google Scholar] [CrossRef]
- Ng, S.K.; Wood, J.P.; Chidlow, G.; Han, G.; Kittipassorn, T.; Peet, D.J.; Casson, R.J. Cancer-like metabolism of the mammalian retina. Clin. Exp. Ophthalmol. 2015, 43, 367–376. [Google Scholar] [CrossRef]
- Galván-Peña, S.; O‘Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Chen, C.; Xiang, A.; Lin, X.; Guo, J.; Liu, J.; Hu, S.; Rui, T.; Ye, Q. Mitophagy: Insights into its signaling molecules, biological functions, and therapeutic potential in breast cancer. Cell Death Discov. 2024, 10, 457. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Chiong, M.; Lavandero, S.; Klionsky, D.J.; Ren, J. Mitophagy in cardiovascular diseases: Molecular mechanisms, pathogenesis, and treatment. Trends Mol. Med. 2022, 28, 836–849. [Google Scholar] [CrossRef]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol. 2017, 7, 170007. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Li, Y.; Xiao, W.; An, W. Alleviation of CCCP-induced mitochondrial injury by augmenter of liver regeneration via the PINK1/Parkin pathway-dependent mitophagy. Exp. Cell Res. 2021, 409, 112866. [Google Scholar] [CrossRef]
- Alam, P.; Maliken, B.D.; Ivey, M.J.; Jones, S.M.; Kanisicak, O. Isolation, Transfection, and Long-Term Culture of Adult Mouse and Rat Cardiomyocytes. J. Vis. Exp. 2020, 164, e61073. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Trempe, J.F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Haile, B.; Arif, M.; Pandey, R.; Rokvic, M.; Nieman, M.; Maliken, B.D.; Paul, A.; Wang, Y.G.; Sadayappan, S.; et al. Inhibition of Senescence-Associated Genes Rb1 and Meis2 in Adult Cardiomyocytes Results in Cell Cycle Reentry and Cardiac Repair Post-Myocardial Infarction. J. Am. Heart Assoc. 2019, 8, e012089. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Huang, W.; Adam, M.; Nieman, M.; Stutenroth, S.; Tranter, M.; Wang, Y.G.; Kanisicak, O. MCM2 mediates post-MI cardioprotection by promoting the pro-angiogenic cardiosome signaling. bioRxiv 2024. [Google Scholar] [CrossRef]
- Elhelaly, W.M.; Lam, N.T.; Hamza, M.; Xia, S.; Sadek, H.A. Redox Regulation of Heart Regeneration: An Evolutionary Tradeoff. Front. Cell Dev. Biol. 2016, 4, 137. [Google Scholar] [CrossRef]
- Bo, B.; Li, S.; Zhou, K.; Wei, J. The Regulatory Role of Oxygen Metabolism in Exercise-Induced Cardiomyocyte Regeneration. Front. Cell Dev. Biol. 2021, 9, 664527. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Gottlieb, R.A. Eat your heart out: Role of autophagy in myocardial ischemia/reperfusion. Autophagy 2008, 4, 416–421. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, Å.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q.; et al. Mitochondrial Cardiomyopathy Caused by Elevated Reactive Oxygen Species and Impaired Cardiomyocyte Proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef]
- Lim, M.L.; Minamikawa, T.; Nagley, P. The protonophore CCCP induces mitochondrial permeability transition without cytochrome c release in human osteosarcoma cells. FEBS Lett. 2001, 503, 69–74. [Google Scholar] [CrossRef]
- Gómez-Sánchez, R.; Gegg, M.E.; Bravo-San Pedro, J.M.; Niso-Santano, M.; Alvarez-Erviti, L.; Pizarro-Estrella, E.; Gutierrez-Martin, Y.; Alvarez-Barrientos, A.; Fuentes, J.M.; González-Polo, R.A.; et al. Mitochondrial impairment increases FL-PINK1 levels by calcium-dependent gene expression. Neurobiol. Dis. 2014, 62, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Vergarajauregui, S.; Wu, C.C.; Piatkowski, T.; Becker, R.; Leone, M.; Hirth, S.; Ricciardi, F.; Falk, N.; Giessl, A.; et al. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. Elife 2015, 4, e05563. [Google Scholar] [CrossRef] [PubMed]
- Engel, F.B.; Schebesta, M.; Duong, M.T.; Lu, G.; Ren, S.; Madwed, J.B.; Jiang, H.; Wang, Y.; Keating, M.T. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005, 19, 1175–1187. [Google Scholar] [CrossRef]
- Pandey, R.; Yang, Y.; Jackson, L.; Ahmed, R.P. MicroRNAs regulating meis1 expression and inducing cardiomyocyte proliferation. Cardiovasc. Regen. Med. 2016, 3, e1468. [Google Scholar]
- Bensley, J.G.; De Matteo, R.; Harding, R.; Black, M.J. Three-dimensional direct measurement of cardiomyocyte volume, nuclearity, and ploidy in thick histological sections. Sci. Rep. 2016, 6, 23756. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adler, C.P.; Friedburg, H.; Herget, G.W.; Neuburger, M.; Schwalb, H. Variability of cardiomyocyte DNA content, ploidy level and nuclear number in mammalian hearts. Virchows. Arch. 1996, 429, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Jiang, Z.; Zagidullin, N.; Liu, T.; Cai, B. Regulation of cardiomyocyte fate plasticity: A key strategy for cardiac regeneration. Signal Transduct. Target. Ther. 2021, 6, 31. [Google Scholar] [CrossRef]
- Malliaras, K.; Terrovitis, J. Cardiomyocyte proliferation vs progenitor cells in myocardial regeneration: The debate continues. Glob. Cardiol. Sci. Pract. 2013, 2013, 303–315. [Google Scholar] [CrossRef]
- da Costa Martins, P.A. Mononuclear Diploidy at the Heart of Cardiomyocyte Proliferation. Cell Stem Cell 2017, 21, 421–422. [Google Scholar] [CrossRef]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, R.P.H.; Kanisicak, O.; Alam, P. Induced Mitophagy Promotes Cell Cycle Re-Entry in Adult Cardiomyocytes. Cells 2025, 14, 853. https://doi.org/10.3390/cells14120853
Ahmed RPH, Kanisicak O, Alam P. Induced Mitophagy Promotes Cell Cycle Re-Entry in Adult Cardiomyocytes. Cells. 2025; 14(12):853. https://doi.org/10.3390/cells14120853
Chicago/Turabian StyleAhmed, Rafeeq P. H., Onur Kanisicak, and Perwez Alam. 2025. "Induced Mitophagy Promotes Cell Cycle Re-Entry in Adult Cardiomyocytes" Cells 14, no. 12: 853. https://doi.org/10.3390/cells14120853
APA StyleAhmed, R. P. H., Kanisicak, O., & Alam, P. (2025). Induced Mitophagy Promotes Cell Cycle Re-Entry in Adult Cardiomyocytes. Cells, 14(12), 853. https://doi.org/10.3390/cells14120853