
Functional Characterization of LTR12C as Regulators of Germ-Cell-Associated TA-p63 in U87-MG and T98-G In Vitro Models
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. IncuCyte® Live-Cell Analysis
2.3. Bioinformatics Analysis
2.4. Total RNA Extraction
2.5. RT-PCR and RT-qPCR Analyses
2.6. SDS-PAGE and Western Blot
2.7. Quantification and Statistical Analysis
3. Results
3.1. DNA Demethylation and HDAC Inhibition Reactivated LTR12C Retroviral Element Expression in GBM Cell Lines
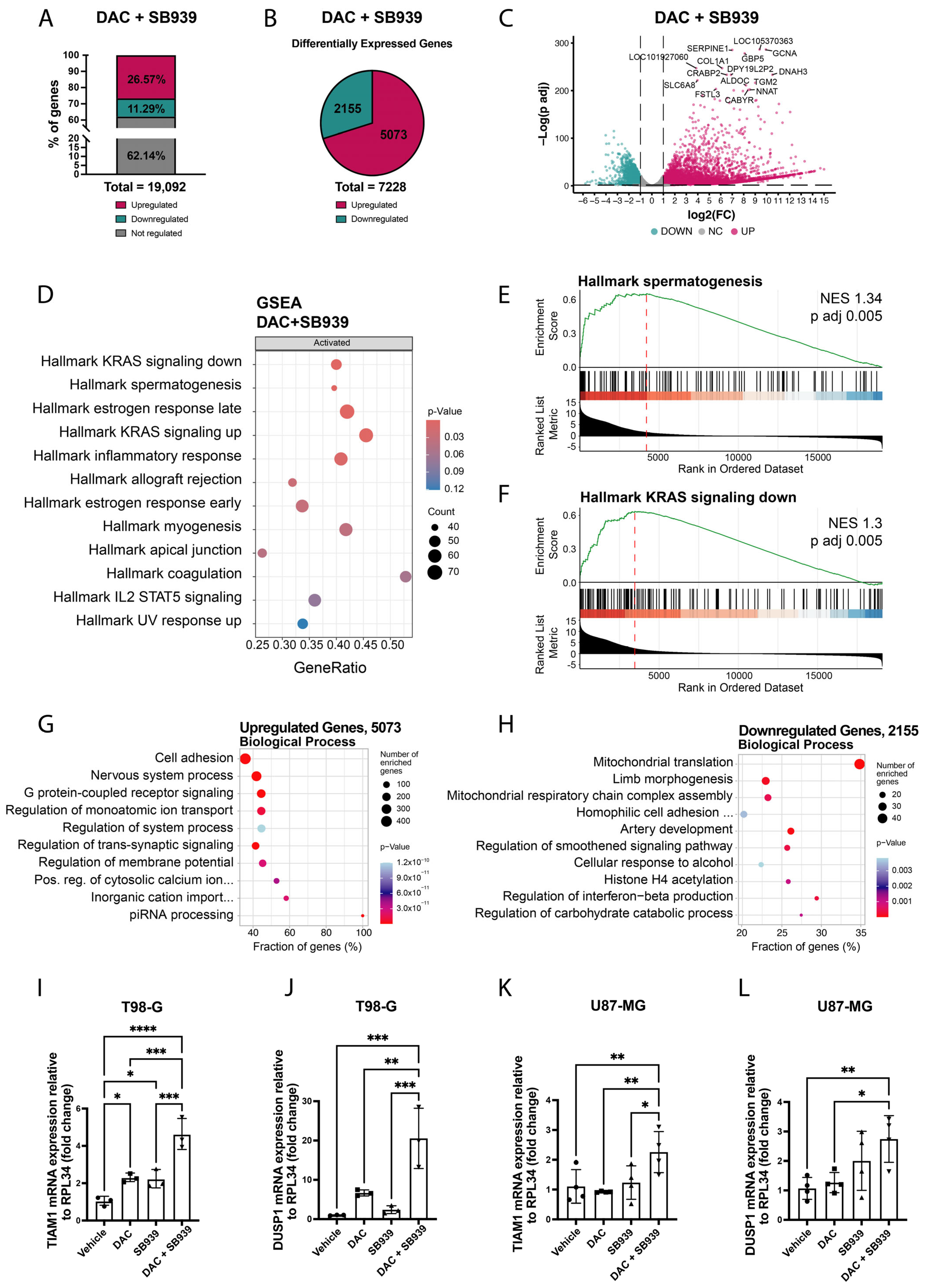
3.2. DNA Demethylation and HDAC Inhibition Induced the Expression of LTR12C-Driven Genes in Glioblastoma Cell Lines
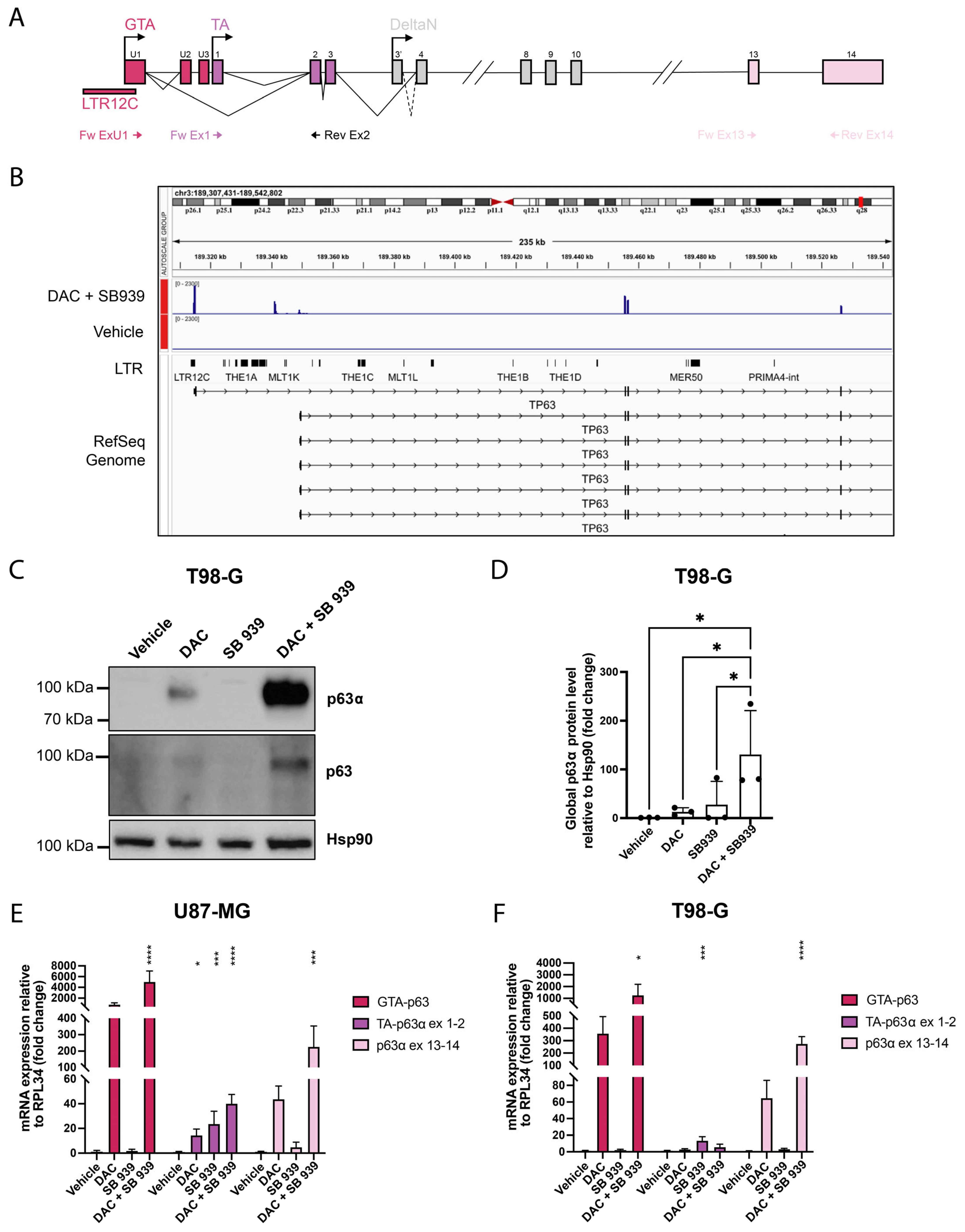
3.2.1. LTR-Driven GTA-p63 Expression in Glioblastoma Cell Models
3.2.2. Characterization of GTA-p63 Transcripts in T98-G Cell Line
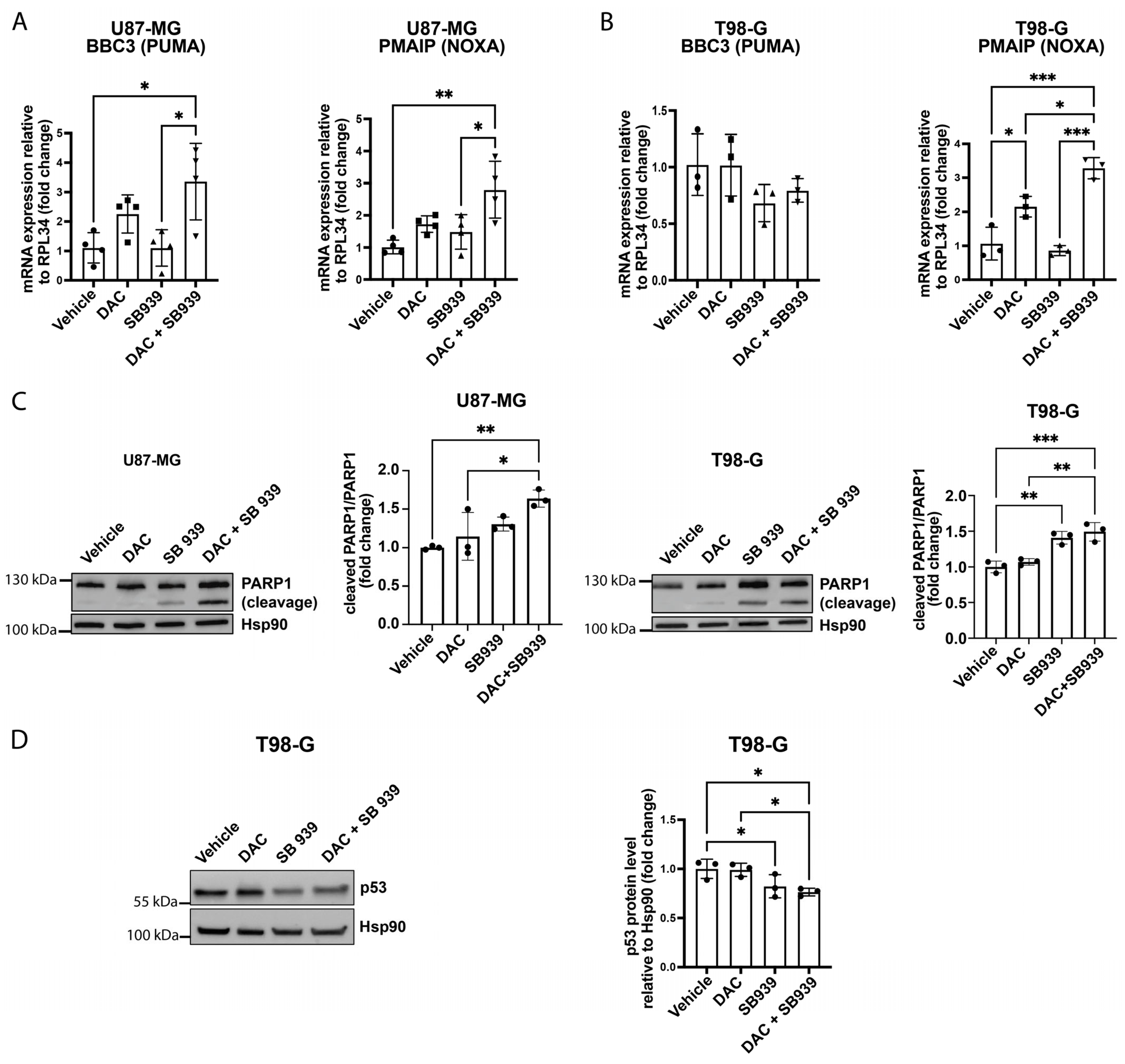
3.3. The Expression of Apoptosis-Related Genes in Glioblastoma Cell Models Could Be Affected by DNMT1 and HDAC Inhibition
3.4. DNA Demethylation and HDAC Inhibition Reduced the Viability of the Glioblastoma Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and Adult Glioblastoma: Multiform (Epi)Genomic Culprits Emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A Review of Glioblastoma Immunotherapy. J. Neurooncol 2021, 151, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E. Origins and Evolutionary Consequences of Ancient Endogenous Retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- Goyal, A.; Bauer, J.; Hey, J.; Papageorgiou, D.N.; Stepanova, E.; Daskalakis, M.; Scheid, J.; Dubbelaar, M.; Klimovich, B.; Schwarz, D.; et al. DNMT and HDAC Inhibition Induces Immunogenic Neoantigens from Human Endogenous Retroviral Element-Derived Transcripts. Nat. Commun. 2023, 14, 6731. [Google Scholar] [CrossRef]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, reviews1017.1. [Google Scholar] [CrossRef]
- Liang, Y.; Qu, X.; Shah, N.M.; Wang, T. Towards Targeting Transposable Elements for Cancer Therapy. Nat. Rev. Cancer 2024, 24, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, X.; Wang, C.; Wang, S.; Jia, L.; Li, L. Endogenous Retroviral Solo-LTRs in Human Genome. Front. Genet. 2024, 15, 1358078. [Google Scholar] [CrossRef]
- Krönung, S.K.; Beyer, U.; Chiaramonte, M.L.; Dolfini, D.; Mantovani, R.; Dobbelstein, M. LTR12 Promoter Activation in a Broad Range of Human Tumor Cells by HDAC Inhibition. Oncotarget 2016, 7, 33484–33497. [Google Scholar] [CrossRef]
- Hu, T.; Pi, W.; Zhu, X.; Yu, M.; Ha, H.; Shi, H.; Choi, J.-H.; Tuan, D. Long Non-Coding RNAs Transcribed by ERV-9 LTR Retrotransposon Act in Cis to Modulate Long-Range LTR Enhancer Function. Nucleic Acids Res. 2017, 45, 4479–4492. [Google Scholar] [CrossRef]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC Inhibitors Induce Cryptic Transcription Start Sites Encoded in Long Terminal Repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef]
- Beyer, U.; Moll-Rocek, J.; Moll, U.M.; Dobbelstein, M. Endogenous Retrovirus Drives Hitherto Unknown Proapoptotic P63 Isoforms in the Male Germ Line of Humans and Great Apes. Proc. Natl. Acad. Sci. USA 2011, 108, 3624–3629. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package, Version 2.56.0; Bioconductor: Seattle, WA, USA, 2024. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.; Hunt, S.E.; Waddell, N.; Spurdle, A.B. A Plugin for the Ensembl Variant Effect Predictor That Uses MaxEntScan to Predict Variant Spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as Designed by Its Users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.; Meola, L.; Lattante, S.; Conte, A.; Sabatelli, M.; Sette, C.; Bernardini, C. Characterization of SOD1-DT, a Divergent Long Non-Coding RNA in the Locus of the SOD1 Human Gene. Cells 2023, 12, 2058. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant graphics for data analysis. Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.12.1; Bioconductor: Seattle, WA, USA, 2021; p. 1. [Google Scholar]
- Blighe, K.; Sharmila, R.; Myles, L. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R Package, version 1.22.0. 2024. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 28 May 2024).
- Bonté, P.-E.; Arribas, Y.A.; Merlotti, A.; Carrascal, M.; Zhang, J.V.; Zueva, E.; Binder, Z.A.; Alanio, C.; Goudot, C.; Amigorena, S. Single-Cell RNA-Seq-Based Proteogenomics Identifies Glioblastoma-Specific Transposable Elements Encoding HLA-I-Presented Peptides. Cell Rep. 2022, 39, 110916. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable Element Expression in Tumors Is Associated with Immune Infiltration and Increased Antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Ohtani, H.; Liu, M.; Liang, G.; Jang, H.J.; Jones, P.A. Efficient Activation of Hundreds of LTR12C Elements Reveals Cis-Regulatory Function Determined by Distinct Epigenetic Mechanisms. Nucleic Acids Res. 2024, 52, 8205–8217. [Google Scholar] [CrossRef] [PubMed]
- Gonfloni, S.; Caputo, V.; Iannizzotto, V. P63 in Health and Cancer. Int. J. Dev. Biol. 2015, 59, 87–93. [Google Scholar] [CrossRef]
- Yu, S.; Yu, X.; Sun, L.; Zheng, Y.; Chen, L.; Xu, H.; Jin, J.; Lan, Q.; Chen, C.C.; Li, M. GBP2 Enhances Glioblastoma Invasion through Stat3/Fibronectin Pathway. Oncogene 2020, 39, 5042–5055. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jin, J.; Zheng, Y.; Zhu, H.; Xu, H.; Ma, J.; Lan, Q.; Zhuang, Z.; Chen, C.C.; Li, M. GBP5 Drives Malignancy of Glioblastoma via the Src/ERK1/2/MMP3 Pathway. Cell Death Dis. 2021, 12, 203. [Google Scholar] [CrossRef]
- Yamaki, T.; Suenaga, Y.; Iuchi, T.; Alagu, J.; Takatori, A.; Itami, M.; Araki, A.; Ohira, M.; Inoue, M.; Kageyama, H.; et al. Temozolomide Suppresses MYC via Activation of TA-p63 to Inhibit Progression of Human Glioblastoma. Sci. Rep. 2013, 3, 1160. [Google Scholar] [CrossRef]
- Lu, J.; Li, S.; Zhang, W.; Zhang, Q.; Liu, C.; Jiang, S.; Xiong, Q.; Meng, X.; Wang, F. ΔNp63 Drives Epithelial Differentiation in Glioma. Clin. Transl. Med. 2020, 10, e165. [Google Scholar] [CrossRef]
- Finlan, L.E.; Hupp, T.R. P63: The Phantom of the Tumor Suppressor. Cell Cycle 2007, 6, 1062–1071. [Google Scholar] [CrossRef]
- Fisher, M.L.; Balinth, S.; Mills, A.A. P63-Related Signaling at a Glance. J. Cell Sci. 2020, 133, jcs228015. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. In Proceedings of the Seventh Annual International Conference on Research in Computational Molecular Biology, Berlin, Germany, 10–14 April 2003; ACM: Berlin, Germany, 2003; pp. 322–331. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Kunadis, E.; Lakiotaki, E.; Korkolopoulou, P.; Piperi, C. Targeting Post-Translational Histone Modifying Enzymes in Glioblastoma. Pharmacol. Ther. 2021, 220, 107721. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. Effect of 5-Aza-2’-Deoxycytidine in Comparison to Valproic Acid and Trichostatin A on Histone Deacetylase 1, DNA Methyltransferase 1, and CIP/KIP Family (P21, P27, and P57) Genes Expression, Cell Growth Inhibition, and Apoptosis Induction in Colon Cancer SW480 Cell Line. Adv. Biomed. Res. 2019, 8, 52. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. P21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Helton, E.S.; Zhang, J.; Chen, X. The Proline-Rich Domain in P63 Is Necessary for the Transcriptional and Apoptosis-Inducing Activities of TA-p63. Oncogene 2008, 27, 2843–2850. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.-C.; Wojcik, B.E.; Huang, H.-J.S.; Friedmann, T.; De Thbolet, N.; Cavenee, W.K. Analysis of the P53 Gene and Its Expression in Human Glioblastoma Cells1. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Hall, C.; Muller, P.A.J. The Diverse Functions of Mutant 53, Its Family Members and Isoforms in Cancer. Int. J. Mol. Sci. 2019, 20, 6188. [Google Scholar] [CrossRef] [PubMed]
- Gebrie, A. Transposable Elements as Essential Elements in the Control of Gene Expression. Mob. DNA 2023, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Kwok, D.W.; Okada, H.; Costello, J.F. Activating the Dark Genome to Illuminate Cancer Vaccine Targets. Nat. Genet. 2024, 56, 1770–1771. [Google Scholar] [CrossRef]
- Andrews, G.; Fan, K.; Pratt, H.E.; Phalke, N.; Consortium, z.; Karlsson, E.K.; Lindblad-Toh, K.; Gazal, S.; Moore, J.E.; Weng, Z.; et al. Mammalian Evolution of Human Cis-Regulatory Elements and Transcription Factor Binding Sites. Science 2023, 380, eabn7930. [Google Scholar] [CrossRef]
- Groh, S.; Schotta, G. Silencing of Endogenous Retroviruses by Heterochromatin. Cell. Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Gao, Y.; Yu, X.-F.; Chen, T. Human Endogenous Retroviruses in Cancer: Expression, Regulation and Function. Oncol. Lett. 2020, 21, 121. [Google Scholar] [CrossRef]
- Zuchegna, C.; Leone, S.; Romano, A.; Porcellini, A.; Messina, S. KRAS Is a Molecular Determinant of Platinum Responsiveness in Glioblastoma. BMC Cancer 2024, 24, 77. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, D.; Reggiardo, R.E.; Lim, J.; Mantalas, G.; Peddu, V.; Kim, D.H. Transposable Element RNA Dysregulation in Mutant KRAS(G12C) 3D Lung Cancer Spheroids. Genomics 2023. [Google Scholar] [CrossRef]
- Scotto, L.; Kinahan, C.; Douglass, E.; Deng, C.; Safari, M.; Casadei, B.; Marchi, E.; Lue, J.K.; Montanari, F.; Falchi, L.; et al. Targeting the T-Cell Lymphoma Epigenome Induces Cell Death, Cancer Testes Antigens, Immune-Modulatory Signaling Pathways. Mol. Cancer Ther. 2021, 20, 1422–1430. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Xu, Y.; Liao, C.; Liu, R.; Liu, J.; Chen, Z.; Zhao, H.; Li, Z.; Chen, L.; Wu, C.; Tan, H.; et al. IRGM Promotes Glioma M2 Macrophage Polarization through P62/TRAF6/NF-κB Pathway Mediated IL-8 Production. Cell Biol. Int. 2019, 43, 125–135. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, R.; Liao, C.; Liu, J.; Zhao, H.; Li, Z.; Liu, W.; Chen, L.; Wu, C.; Tan, H.; et al. High Expression of Immunity-Related GTPase Family M Protein in Glioma Promotes Cell Proliferation and Autophagy Protein Expression. Pathol.-Res. Pract. 2019, 215, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Grespi, F.; Annicchiarico-Petruzzelli, M.; Melino, G. P63 the Guardian of Human Reproduction. Cell Cycle 2012, 11, 4545–4551. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, X.; Xiong, Q.; Han, J.; Zhu, Q. The Dual Role of P63 in Cancer. Front. Oncol. 2023, 13, 1116061. [Google Scholar] [CrossRef]
- Jang, H.J.; Shah, N.M.; Maeng, J.H.; Liang, Y.; Basri, N.L.; Ge, J.; Qu, X.; Mahlokozera, T.; Tzeng, S.-C.; Williams, R.B.; et al. Epigenetic Therapy Potentiates Transposable Element Transcription to Create Tumor-Enriched Antigens in Glioblastoma Cells. Nat. Genet. 2024, 56, 1903–1913. [Google Scholar] [CrossRef]
- Pitzius, S.; Osterburg, C.; Gebel, J.; Tascher, G.; Schäfer, B.; Zhou, H.; Münch, C.; Dötsch, V. TA*p63 and GTA-p63 Achieve Tighter Transcriptional Regulation in Quality Control by Converting an Inhibitory Element into an Additional Transactivation Domain. Cell Death Dis. 2019, 10, 686. [Google Scholar] [CrossRef]
- Sawa, H.; Murakami, H.; Kumagai, M.; Nakasato, M.; Yamauchi, S.; Matsuyama, N.; Tamura, Y.; Satone, A.; Ide, W.; Hashimoto, I.; et al. Histone Deacetylase Inhibitor, FK228, Induces Apoptosis and Suppresses Cell Proliferation of Human Glioblastoma Cells in Vitro and in Vivo. Acta Neuropathol. 2004, 107, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic Modulation and Understanding of HDAC Inhibitors in Cancer Therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Khan, S.; Kumar, S.; Sinha, S.; Farhan, M.; Bora, H.K.; Maurya, R.; Meeran, S.M. Cucurbitacin B Alters the Expression of Tumor-Related Genes by Epigenetic Modifications in NSCLC and Inhibits NNK-Induced Lung Tumorigenesis. Cancer Prev. Res. 2015, 8, 552–562. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Montalban-Bravo, G.; Berdeja, J.G.; Abaza, Y.; Jabbour, E.; Essell, J.; Lyons, R.M.; Ravandi, F.; Maris, M.B.; Heller, B.; et al. Phase 2, Randomized, Double-Blind Study of Pracinostat in Combination with Azacitidine in Patients with Untreated, Higher-Risk Myelodysplastic Syndromes. Cancer 2017, 123, 994–1002. [Google Scholar] [CrossRef]
- Whitmore, M.A.; Li, H.; Lyu, W.; Khanam, S.; Zhang, G. Epigenetic Regulation of Host Defense Peptide Synthesis: Synergy between Histone Deacetylase Inhibitors and DNA/Histone Methyltransferase Inhibitors. Front. Immunol. 2022, 13, 874706. [Google Scholar] [CrossRef]
- Blagitko-Dorfs, N.; Schlosser, P.; Greve, G.; Pfeifer, D.; Meier, R.; Baude, A.; Brocks, D.; Plass, C.; Lübbert, M. Combination Treatment of Acute Myeloid Leukemia Cells with DNMT and HDAC Inhibitors: Predominant Synergistic Gene Downregulation Associated with Gene Body Demethylation. Leukemia 2018, 33, 945–956. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (nM) | U87-MG | T98-G |
|---|---|---|
| DAC | 332 ± 97 | 248 ± 35 |
| SB939 | 1327 ± 74 | 1048 ± 136 |
| DAC + SB939 | 406 ± 133 | 175 ± 42 |
| Bliss Score (Combination Index) | −1472 | −1082 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meola, L.; Shetty, S.R.; Peschiaroli, A.; Sette, C.; Bernardini, C. Functional Characterization of LTR12C as Regulators of Germ-Cell-Associated TA-p63 in U87-MG and T98-G In Vitro Models. Cells 2025, 14, 852. https://doi.org/10.3390/cells14110852
Meola L, Shetty SR, Peschiaroli A, Sette C, Bernardini C. Functional Characterization of LTR12C as Regulators of Germ-Cell-Associated TA-p63 in U87-MG and T98-G In Vitro Models. Cells. 2025; 14(11):852. https://doi.org/10.3390/cells14110852
Chicago/Turabian StyleMeola, Lucia, Sohum Rajesh Shetty, Angelo Peschiaroli, Claudio Sette, and Camilla Bernardini. 2025. "Functional Characterization of LTR12C as Regulators of Germ-Cell-Associated TA-p63 in U87-MG and T98-G In Vitro Models" Cells 14, no. 11: 852. https://doi.org/10.3390/cells14110852
APA StyleMeola, L., Shetty, S. R., Peschiaroli, A., Sette, C., & Bernardini, C. (2025). Functional Characterization of LTR12C as Regulators of Germ-Cell-Associated TA-p63 in U87-MG and T98-G In Vitro Models. Cells, 14(11), 852. https://doi.org/10.3390/cells14110852