



Defective Mitochondrial Respiration in Hereditary Thoracic Aneurysms

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Procedures
2.2. Lentivirus Production and Infection
2.3. Extracellular Flux Analysis and Metabolic Assays
2.4. Gelatin Zymography
2.5. Quantitative PCR
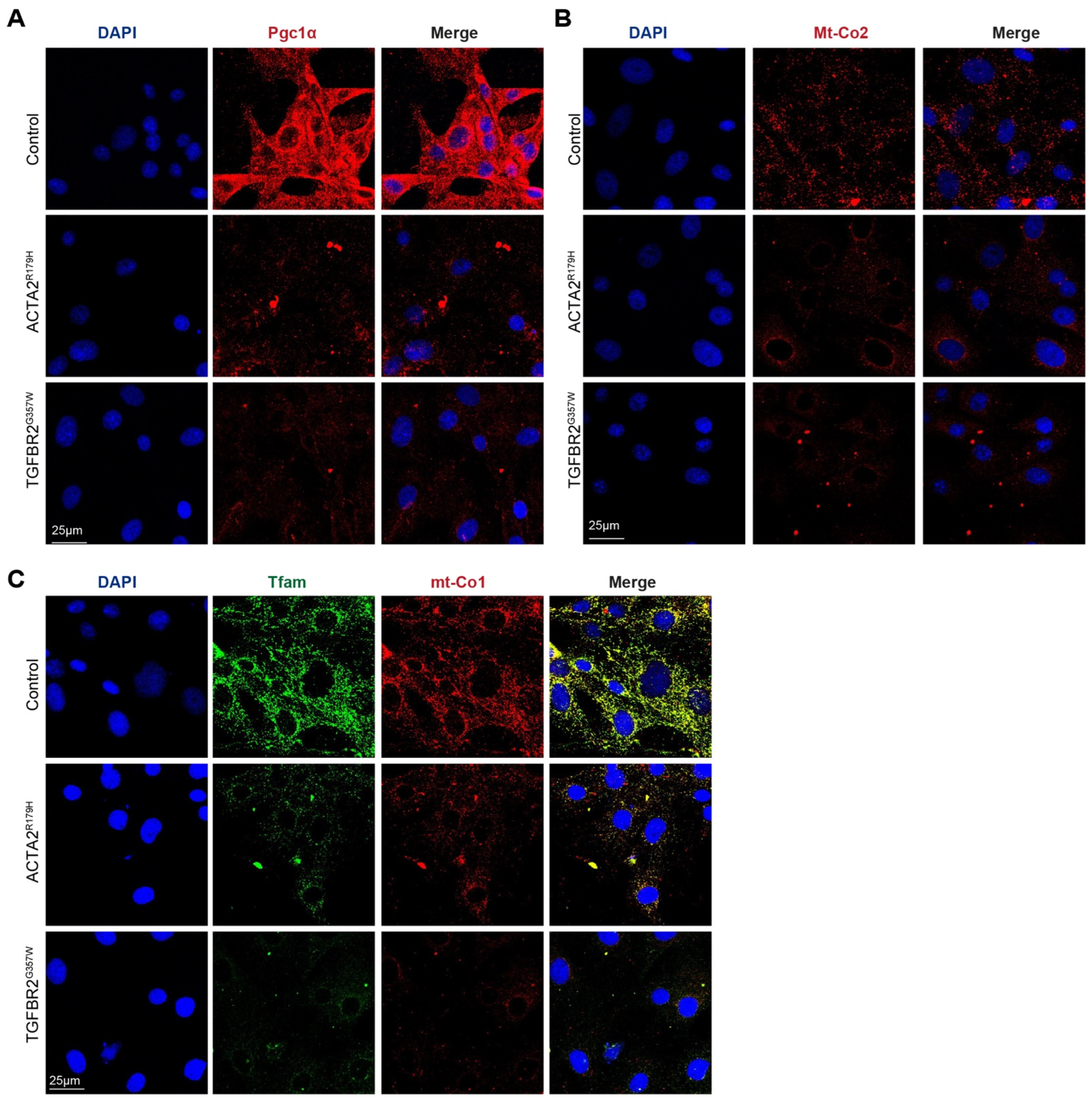
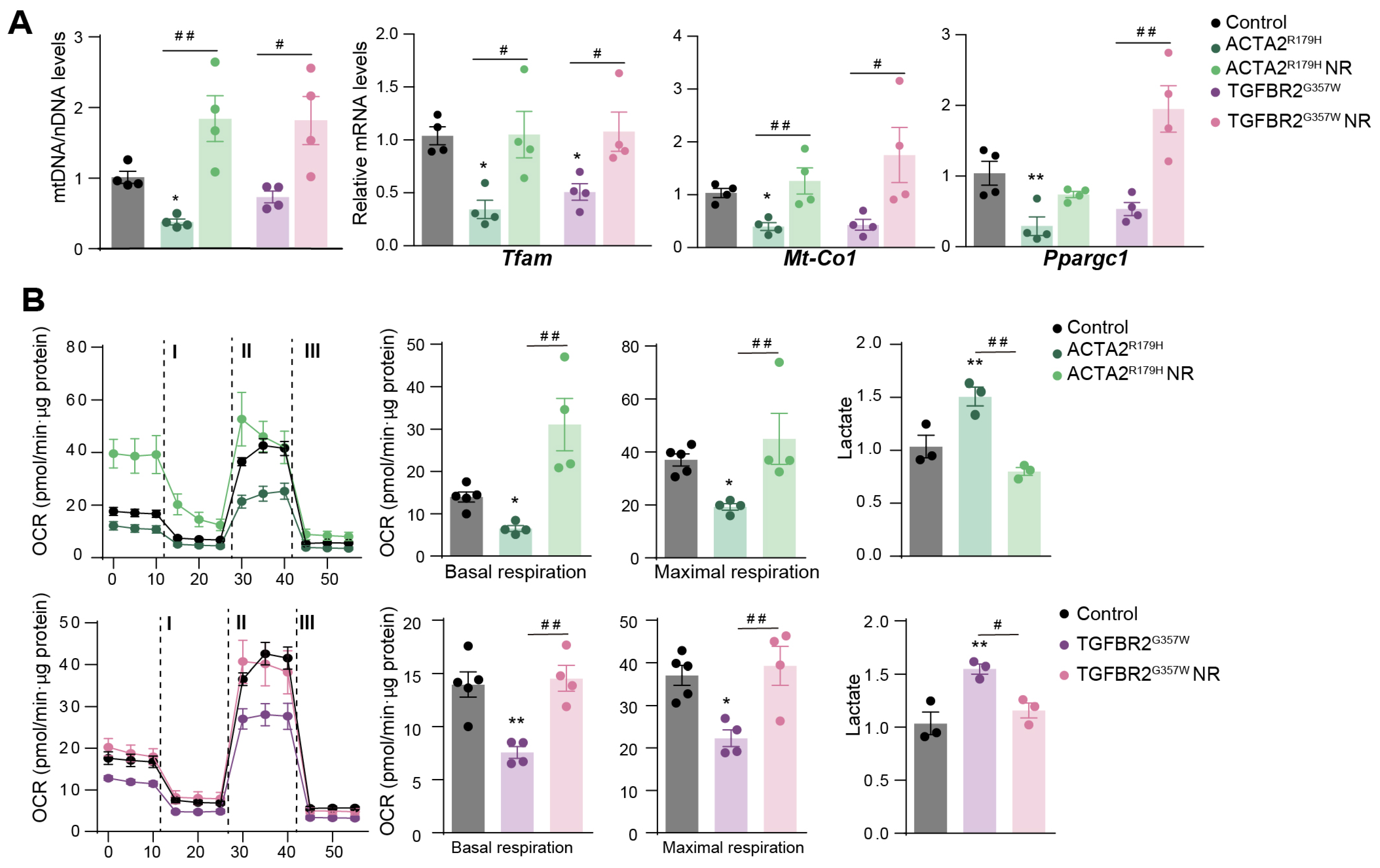
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Rylski, B.; Schilling, O.; Czerny, M. Acute aortic dissection: Evidence, uncertainties, and future therapies. Eur. Heart J. 2023, 44, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, S.M.; Sloan, B.; Jones, J.A. Pathophysiology and Pathogenesis of Marfan Syndrome. In Progress in Heritable Soft Connective Tissue Diseases; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; pp. 185–206. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural history of thoracic aortic aneurysms. J. Vasc. Surg. 2012, 56, 565–571. [Google Scholar] [CrossRef]
- Benke, K.; Ágg, B.; Szilveszter, B.; Tarr, F.; Nagy, Z.B.; Pólos, M.; Daróczi, L.; Merkely, B.; Szabolcs, Z. The role of transforming growth factor-beta in Marfan syndrome. Cardiol. J. 2013, 20, 227–234. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Roldan-Montero, R.; Ruiz-Rodríguez, M.J.; Redondo, J.M.; Martín-Ventura, J.L.; Mittelbrunn, M. Rewiring Vascular Metabolism Prevents Sudden Death due to Aortic Ruptures—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 462–469. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Ruiz-Rodríguez, M.J.; Desdín-Micó, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuña, P.; et al. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Oller, J.; Méndez-Barbero, N.; Ruiz, E.J.; Villahoz, S.; Renard, M.; Canelas, L.I.; Briones, A.M.; Alberca, R.; Lozano-Vidal, N.; Hurlé, M.A.; et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med. 2017, 23, 200–212. [Google Scholar] [CrossRef]
- Lino Cardenas, C.L.; Kessinger, C.W.; Cheng, Y.; MacDonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Yeri, A.S.; Jaffer, F.A.; et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1009. [Google Scholar] [CrossRef]
- Nguyen, T.A.V.; Lino, C.A.; Hang, H.T.; Alves, J.V.; Thang, B.Q.; Shin, S.J.; Sugiyama, K.; Matsunaga, H.; Takeyama, H.; Yamashiro, Y.; et al. Protective Role of Endothelial Fibulin-4 in Valvulo-Arterial Integrity. J. Am. Heart Assoc. 2023, 12, e026942. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Ravi, S.; Xie, N.; Persons, B.P.; Rangarajan, S.; Zmijewski, J.W.; Mitra, K.; Liu, G.; Darley-Usmar, V.M.; et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem. 2015, 290, 25427–25438. [Google Scholar] [CrossRef]
- Kim, J.; Cheong, J.-H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells 2020, 9, 1691. [Google Scholar] [CrossRef]
- Branchetti, E.; Poggio, P.; Sainger, R.; Shang, E.; Grau, J.B.; Jackson, B.M.; Lai, E.K.; Parmacek, M.S.; Gorman, R.C.; Gorman, J.H.; et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc. Res. 2013, 100, 316–324. [Google Scholar] [CrossRef]
- Irace, F.G.; Cammisotto, V.; Valenti, V.; Forte, M.; Schirone, L.; Bartimoccia, S.; Iaccarino, A.; Peruzzi, M.; Schiavon, S.; Morelli, A.; et al. Role of Oxidative Stress and Autophagy in Thoracic Aortic Aneurysms. JACC Basic Transl. Sci. 2021, 6, 719–730. [Google Scholar] [CrossRef]
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125. [Google Scholar] [CrossRef]
- Zhang, H.; Tsui, C.K.; Garcia, G.; Joe, L.K.; Wu, H.; Maruichi, A.; Fan, W.; Pandovski, S.; Yoon, P.H.; Webster, B.M.; et al. The extracellular matrix integrates mitochondrial homeostasis. Cell 2024, 187, 4289–4304.e26. [Google Scholar] [CrossRef]
- van der Pluijm, I.; Burger, J.; van Heijningen, P.M.; IJpma, A.; van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.-J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Wan, S.; Qi, J.; Hao, Y.; An, P.; Luo, Y.; Luo, J. Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown. Int. J. Mol. Sci. 2024, 25, 2662. [Google Scholar] [CrossRef]
- Hibender, S.; Franken, R.; Van Roomen, C.; Ter Braake, A.; Van Der Made, I.; Schermer, E.E.; Gunst, Q.; Van Den Hoff, M.J.; Lutgens, E.; Pinto, Y.M.; et al. Resveratrol Inhibits Aortic Root Dilatation in the Fbn1 C1039G/+ Marfan Mouse Model. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Marcos-Ríos, D.; Rochano-Ortiz, A.; San Sebastián-Jaraba, I.; Fernández-Gómez, M.J.; Méndez-Barbero, N.; Oller, J. Mitochondrial Dysfunction: A New Hallmark in Hereditable Thoracic Aortic Aneurysm Development. Cells 2025, 14, 618. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcos-Ríos, D.; Rochano-Ortiz, A.; Méndez-Barbero, N.; Oller, J. Defective Mitochondrial Respiration in Hereditary Thoracic Aneurysms. Cells 2025, 14, 768. https://doi.org/10.3390/cells14110768
Marcos-Ríos D, Rochano-Ortiz A, Méndez-Barbero N, Oller J. Defective Mitochondrial Respiration in Hereditary Thoracic Aneurysms. Cells. 2025; 14(11):768. https://doi.org/10.3390/cells14110768
Chicago/Turabian StyleMarcos-Ríos, Daniel, Antonio Rochano-Ortiz, Nerea Méndez-Barbero, and Jorge Oller. 2025. "Defective Mitochondrial Respiration in Hereditary Thoracic Aneurysms" Cells 14, no. 11: 768. https://doi.org/10.3390/cells14110768
APA StyleMarcos-Ríos, D., Rochano-Ortiz, A., Méndez-Barbero, N., & Oller, J. (2025). Defective Mitochondrial Respiration in Hereditary Thoracic Aneurysms. Cells, 14(11), 768. https://doi.org/10.3390/cells14110768