
Perspectives of Targeting Autophagy as an Adjuvant to Anti-PD-1/PD-L1 Therapy for Colorectal Cancer Treatment


Abstract

1. Introduction
2. Overview of PD-1/PD-L1
2.1. PD-1/PD-L1 in CRC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MSI-H/dMMR CRC Phenotype | Immune Target | Treatment Combination | Study Phase | Clinical Trial Identifier | Status | Outcome |
|---|---|---|---|---|---|---|
| mCRC MSI-H/dMMR | PD-1 | Pembrolizumab | II | NCT02460198 | Completed | ORR = 32.8% [28,33,34] |
| MSI-H/dMMR tumors | PD-1 | Pembrolizumab | II | NCT01876511 | Completed | (ORR) = 50% [27,30] |
| MSI-H/dMMR CRC | PD-1 | Pembrolizumab + Epacadosat | I/II | NCT02178722 | Completed | ORR(I) = 57% ORR(II) = 80% |
| Advanced (CRC) | PD-1 | Varlilumab + Nivolumab | I II | NCT02335918 | Completed | ORR = 5% [29] |
| MSI-H/dMMR and MSS/pMMR mCRC and MSI-H/dMMR endometrial carcinoma | PD-1 | Pembrolizumab + Ataluren | I-II | NCT04014530 | Recruiting | ORR = 71% [30] |
| MSI-H/dMMR CRC | PD-1 | Pembrolizumab + COX inhibitor (aspirin) | II | NCT03638297 | Recruiting | N.A. |
| MSI-H/dMMRmetastatic solid tumors | PD-1 | Pembrolizumab + RT (metastatic site) vs. Pembrolizumab | II | NCT04001101 | Recruiting | N.A. |
| MSI-H/dMMR mCRC | PD-1 | Nivolumab + Ipilimumab | II | NCT04730544 | Recruiting | N.A. |
| MSI-H/dMMR mCRC | PD-L1 | Avelumab | II | NCT03186326 | Recruiting | N.A. |
| Advanced or metastatic solid tumors, including MSI-H/dMMR CRC | PD-L1 | Avelumab + Regorafenib | I-II | NCT03475953 | Recruiting | N.A. |
| MSI-H/dMMR or POLE mutated mCRC | PD-L1 | Durvalumab | II | NCT03435107 | Active, not recruiting | N.A. |
| Advanced pancreatic cancer NSCLC dMMR CRC | PD-L1 | Danvatirsen + Durvalumab | II | NCT02983578 | Active, not recruiting | N.A. |
| Advanced MSI-H/dMMR CRC | PD-L1 | Durvalumab | II | NCT02227667 | Completed | N.A. |
| Metastatic/ advanced CRC and PaC | PD-L1 | Durvalumab + Pexidartinib | I | NCT02777710 | Completed | N.A. |
| Advanced chemotherapy-resistant MSI/dMMR CRC | PD-L1 | Atezolizumab + Bevacizumab | II | NCT02982694 | Recruiting | N.A. |
| MSI-H/dMMR mCRC | PD-L1 | Atezolizumab vs. Atezolizumab + Bevacizumab + FOLFOX | III | NCT02997228 | Recruiting | N.A. |
| MSI-H/dMMR mCRC | PD-1 + CTLA-4 | Nivolumab vs. Nivolumab + Ipilimumab Nivolumab + Ipilimumab vs. chemotherapy | III | NCT04008030 | Recruiting | N.A. |
| MSI-H/dMMR CRC, MMS CRC, pancreatic cancer | PD-1 + CTLA-4 | Nivolumab + Ipilimumab + RT | II | NCT03104439 | Recruiting | N.A. |
| Recurrent or metastatic MSI-H and non-MSI-H CRC | PD-1 + CTLA-4 | Nivolumab Nivolumab + Ipilimumab Nivolumab + Ipiliumab + Cobimetinib Nivolumab + BMS-986016 Nivolumab + Daratumumab | II | NCT02060188 | Active, not recruiting | N.A. |
| MMS/pMMR CRC Phenotype | Immune Target/Generic Name | Treatment Combination | Study Phase | Clinical Trial Identifier | Status | Outcome |
|---|---|---|---|---|---|---|
| advanced MSS/pMMR mCRC | PD-L1 + PD-1 | Regorafenib + Nivolumab | II | NCT04126733 | Completed | ORR = 33% [35] |
| mCRC and pancreatic cancer | PD-1 | Olaptesed pegol + Pembrolizumab | I/II | NCT03168139 | Completed | N.A. |
| Advanced MSS/pMMR CRC | PD-1 | Pembrolizumab + cyclophosphamide + Colon cancer vaccine | II | NCT02981524 | Completed | ORR = 1.6% Did not meet its primary objective in MSS/pMMR CRC [36] |
| Refractory MSS/ pMMR mCRC | PD-1 | Pembrolizumab + Maraviroc | I | NCT03274804 | Completed | Therapy combination is feasible with a beneficial toxicity pattern [37] |
| MSS/pMMR CRC | PD-1 | Nivolumab + Tipiracil hydrochloride | II | NCT02860546 | Completed | Therapy combination is feasibly tolerable. No clinical benefit to MSS, mCRC failed [38] |
| MSS/pMMR CRC | PD-L1 | Avelumab + Tomivosertib vs. Tomivosertib | II | NCT03258398 | Completed | N.A. |
| Chemorefractory MSS/pMMR mCRC | PD-1 | Pembrolizumab + Azacitidine | II | NCT02260440 | Completed | ORR = 3% The therapy combination is safe and tolerable with modest clinical activity [39] |
| MSS/pMMR mCRC mPaC | PD-1 | Olaptesed pegol vs. Olaptesed pegol + Pembrolizumab | I/II | NCT03168139 | Completed | N.A. |
| Advanced solid tumors (including MSS/pMMR CRC) | PD-L1 | Azacitidine + Durvalumab | II | NCT02811497 | Completed | Did not show strong effects in immunologically cold solid tumors [32] |
| MSS/pMMR mCRC (Liver) | PD-L1 | Durvalumab + Tremelimumab following radioembolization (RE) with SIR-spheres | I | NCT03005002 | Completed | N.A. |
| Non MSI-H mCRC | PD-L1 | Cobimetinib + Bevacizumab + Atezolizumab | Ib | NCT02876224 | Completed | N.A. |
| mCRC | PD-L1 | Atezolizumab (A) vs. Atezolizumab (A) + Cobimetinib (C) vs. Regorafenib | III | NCT02788279 | Completed | Therapy combination (B and C) did not improve overall survival. Safety of the therapy combination is consistent with the individual drugs |
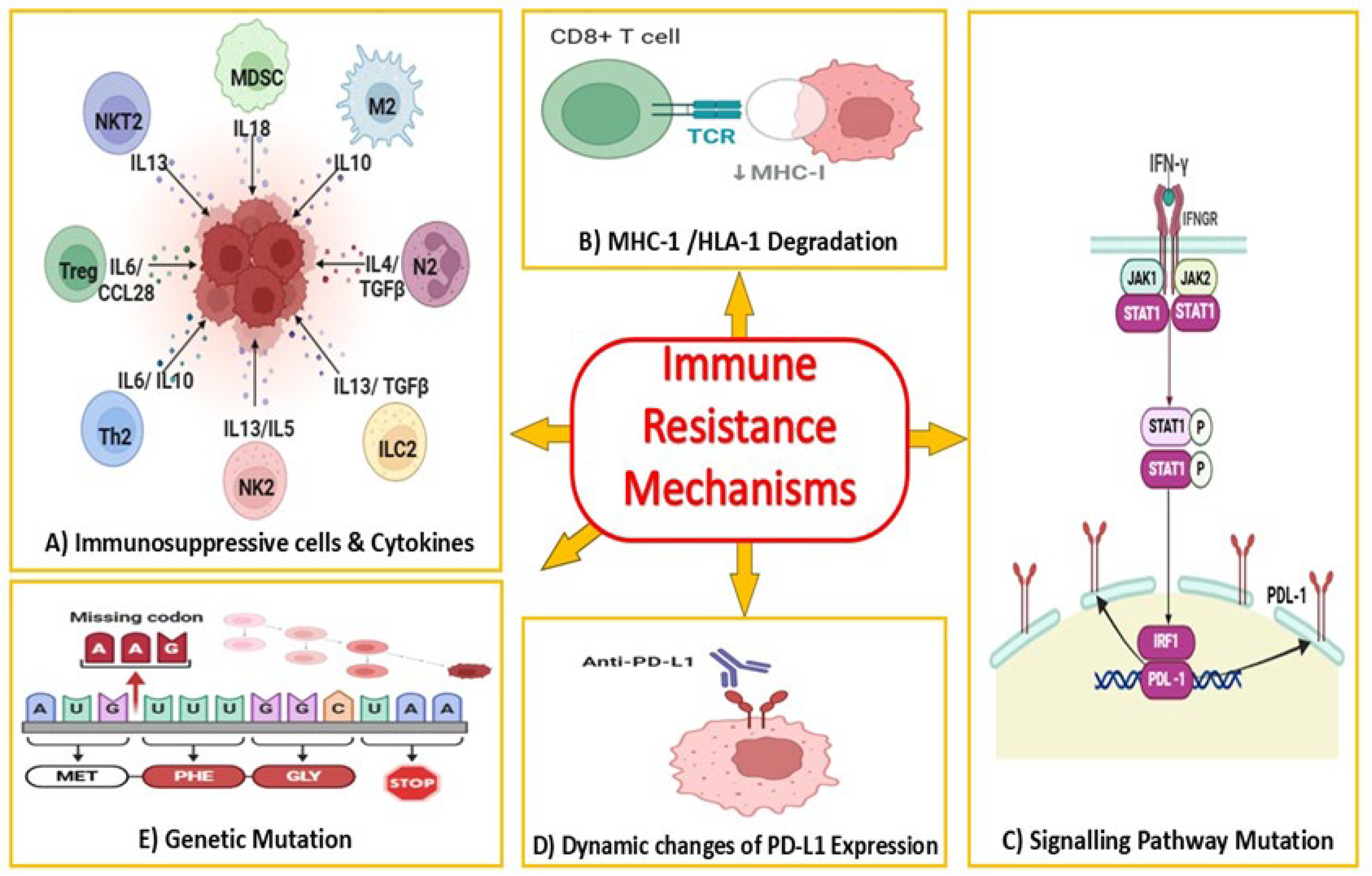
2.2. Current Limitations of Anti-PD-1/PD-L1 Therapy in Colorectal Cancer
3. Autophagy as a Modulator of Immune Response in Cancer
3.1. The Role of Autophagy in the Tumor Microenvironment
3.2. Autophagy and Antigen Presentation
3.3. Tumor Cell Autophagy
| Cancer | Drug Resistance Mediated by Autophagy Induction | Autophagy Inhibitor | Mechanism of Targeting Drug-Resistant Cancer Cells | Reference |
|---|---|---|---|---|
| Colorectal Cancer | PFKFB3 inhibitor, 3PO | 3-methyladenine/Chloroquine | Inhibition of autophagy induced due to PFKFB3 inhibition | [114] |
| Colorectal Cancer | Cabozantinib XL184 | SBI0226365/Chloroquine | Inhibition of autophagy-dependent metabolism | [112] |
| Colon Cancer | Inhibition of ANKRD37 | Chloroquine | Inhibition of autophagy is induced due to ANKRD37 translocation to the nucleus | [113] |
| Colon Cancer Cells | CoCl2 | 3-methyladenine | Inhibits hypoxia-induced autophagy | [105] |
| Colon Cancer | NA | 3-methyladenine | Inhibits the supply of free fatty acid (FFA) from adipocytes | [111] |
| Colon adenocarcinoma | Oxaliplatin | SP600125 | JNK inhibition prevents hypoxia-induced autophagy | [110] |
3.4. Autophagy in Immune Cells and Others
4. Preclinical Models for Testing Anti-PD-1/PD-L1 Therapies
4.1. Clinical Outcome for Anti-PD-1/PD-L1 Using Different Models
4.2. Study of Autophagy in Animal Models
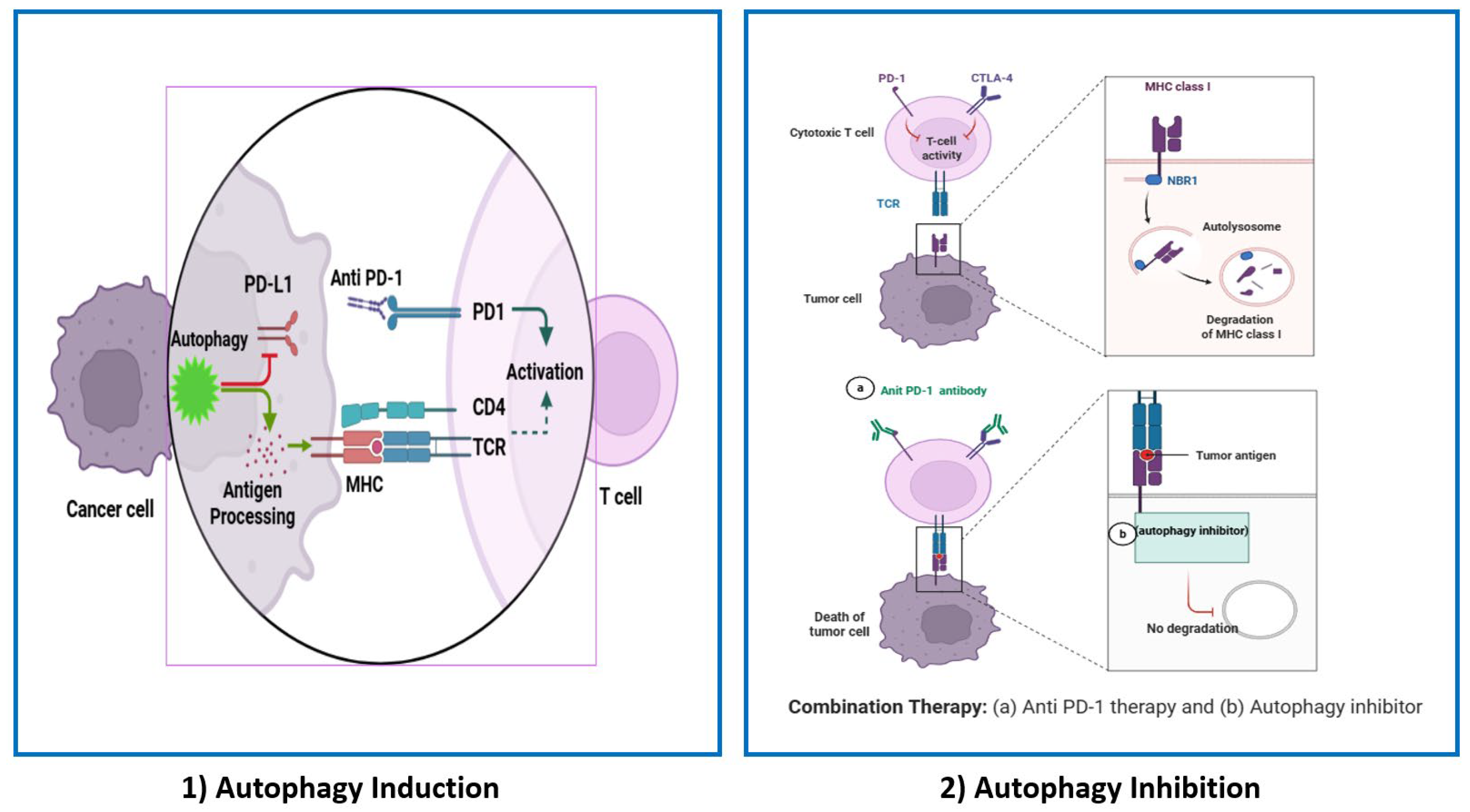
5. Therapeutic Perspectives of Targeting Autophagy to Enhance the Response of PD-1/PD-L1 Therapy
| Tumor Types | Agent | Modulation | Related Mechanisms | Outcome | Reference |
|---|---|---|---|---|---|
| CRC | CXCL1 | autophagy induction | reduce MHC-I expression | immune inhibition | [160] |
| CRC | Brucine | autophagy inhibition | enhance calreticulin and HMGB1 release | immune activation | [161] |
| CRC | FuFangChangTai Decoction | autophagy induction | activate macrophages and increase expression of CD86 and CD40 | immune activation | [162] |
| CRC | Zosuquidar | PD-L1 selective autophagy | reduce PD-L1 expression | immune activation | [163] |
| CRC | Rigosertib | PD-L1 selective autophagy | reduce PD-L1 expression | immune activation | [164] |
| CRC | Nod1 | Autophagy induction | M2 polarization | immune inhibition | [165] |
| Target | Autophagy Inhibitor | Study Type | Clinical Trial Identifier | Outcome | References |
|---|---|---|---|---|---|
| PI3Kinase inhibition | Copanlisib + Nivolumab | Clinical Trial I/II | NCT03711058 | Recruiting N.A. | [166] |
| Inhibition of VPS34 | SB02024/SAR405 | In vivo | - | Enhanced antitumor efficacy | [167] |
| Lysosomes, PPT1 | Hydroxychloroquine + anti-PD-1 | In vivo | - | Tumor growth impairment and improved survival in mouse models | [156] |
6. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kanth, P.; Inadomi, J.M. Screening and prevention of colorectal cancer. BMJ 2021, 374, n1855. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Wang, H.; Tian, T.; Zhang, J. Tumor-Associated Macrophages (TAMs) in Colorectal Cancer (CRC): From Mechanism to Therapy and Prognosis. Int. J. Mol. Sci. 2021, 22, 8470. [Google Scholar] [CrossRef]
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr. Drug Targets 2021, 22, 998–1009. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and emerging therapeutic approaches for colorectal cancer: A comprehensive review. World J. Gastrointest. Surg. 2023, 15, 495–519. [Google Scholar] [CrossRef]
- Morris, V.K.; Kennedy, E.B.; Baxter, N.N.; Benson, A.B.; Cercek, A.; Cho, M.; Ciombor, K.K.; Cremolini, C.; Davis, A.; Deming, D.A.; et al. Treatment of Metastatic Colorectal Cancer: ASCO Guideline. J. Clin. Oncol. 2023, 41, 678–700. [Google Scholar] [CrossRef]
- Brown, L.J.; Da Silva, I.P.; Moujaber, T.; Gao, B.; Hui, R.; Gurney, H.; Carlino, M.; Nagrial, A. Five-year survival and clinical correlates among patients with advanced non-small cell lung cancer, melanoma and renal cell carcinoma treated with immune checkpoint inhibitors in Australian tertiary oncology centres. Cancer Med. 2023, 12, 6788–6801. [Google Scholar] [CrossRef]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of Programmed Death 1 Ligands by Murine T Cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Cervantes, B.; André, T.; Cohen, R. Deficient mismatch repair/microsatellite unstable colorectal cancer: Therapeutic advances and questions. Ther. Adv. Med. Oncol. 2024, 16, 17588359231170473. [Google Scholar] [CrossRef]
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1624. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, C.; Chen, M.; Jiang, Y.; Wu, Y.; Mao, R.; Fan, Y. The generation of PD-L1 and PD-L2 in cancer cells: From nuclear chromatin reorganization to extracellular presentation. Acta Pharm. Sin. B 2022, 12, 1041–1053. [Google Scholar] [CrossRef]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X. Effects of molecular markers on the treatment decision and prognosis of colorectal cancer: A narrative review. J. Gastrointest. Oncol. 2021, 12, 1191–1196. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Motta, R.; Cabezas-Camarero, S.; Torres-Mattos, C.; Riquelme, A.; Calle, A.; Figueroa, A.; Sotelo, M.J. Immunotherapy in microsatellite instability metastatic colorectal cancer: Current status and future perspectives. J. Clin. Transl. Res. 2021, 7, 511–522. [Google Scholar]
- Kiran, N.S.; Yashaswini, C.; Maheshwari, R.; Bhattacharya, S.; Prajapati, B.G. Advances in Precision Medicine Approaches for Colorectal Cancer: From Molecular Profiling to Targeted Therapies. ACS Pharmacol. Transl. Sci. 2024, 7, 967–990. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Chen, X.; Chen, L.-J.; Peng, X.-F.; Deng, L.; Wang, Y.; Li, J.-J.; Guo, D.-L.; Niu, X.-H. Anti-PD-1/PD-L1 therapy for colorectal cancer: Clinical implications and future considerations. Transl. Oncol. 2024, 40, 101851. [Google Scholar] [CrossRef]
- Baimas-George, M.; Baker, E.; Kamionek, M.; Salmon, J.S.; Sastry, A.; Levi, D.; Vrochides, D. A Complete Pathological Response to Pembrolizumab following ex vivo Liver Resection in a Patient with Colorectal Liver Metastases. Chemotherapy 2018, 63, 90–94. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, J.; Deng, Y.; Wang, H. Complete response in patients with locally advanced rectal cancer after neoadjuvant treatment with nivolumab. OncoImmunology 2019, 8, e1663108. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Van Den Berg, J.G.; Beets, G.L.; Lopez-Yurda, M.; Aalbers, A.G.; Grootscholten, C.; Snaebjornsson, P.; Maas, M.; Mertz, M.; et al. Neoadjuvant ipilimumab plus nivolumab in early-stage colon cancer. Ann. Oncol. 2018, 29, viii731. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability–High/Mismatch Repair–Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Sanborn, R.E.; Pishvaian, M.J.; Callahan, M.K.; Weise, A.; Sikic, B.I.; Rahma, O.; Cho, D.C.; Rizvi, N.A.; Sznol, M.; Lutzky, J.; et al. Safety, tolerability and efficacy of agonist anti-CD27 antibody (varlilumab) administered in combination with anti-PD-1 (nivolumab) in advanced solid tumors. J. Immunother. Cancer 2022, 10, e005147. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Thomas, J.; Leal, A.; Overman, M.J. Clinical Development of Immunotherapy for Deficient Mismatch Repair Colorectal Cancer. Clin. Color. Cancer 2020, 19, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.; Loo Yau, H.; Chakravarthy, A.; Wang, B.; Shen, S.Y.; Ettayebi, I.; Ishak, C.A.; Bedard, P.L.; Abdul Razak, A.; RHansen, A.R.; et al. An open-label, phase II multicohort study of an oral hypomethylating agent CC-486 and durvalumab in advanced solid tumors. J. Immunother. Cancer 2020, 8, e000883. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Diaz, L.A.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.H.; Kavan, P.; et al. Pembrolizumab for previously treated, microsatellite instability–high/mismatch repair–deficient advanced colorectal cancer: Final analysis of KEYNOTE-164. Eur. J. Cancer 2023, 186, 185–195. [Google Scholar] [CrossRef]
- van Vugt, M.J.; Stone, J.A.; De Greef, R.H.; Snyder, E.S.; Lipka, L.; Turner, D.C.; Chain, A.; Lala, M.; Li, M.; Robey, S.H.; et al. Immunogenicity of pembrolizumab in patients with advanced tumors. J. Immunother. Cancer 2019, 7, 212. [Google Scholar] [CrossRef]
- Fakih, M.; Raghav, K.P.S.; Chang, D.Z.; Larson, T.; Cohn, A.L.; Huyck, T.K.; Cosgrove, D.; Fiorillo, J.A.; Tam, R.; D’Adamo, D.; et al. Regorafenib plus nivolumab in patients with mismatch repair-proficient/microsatellite stable metastatic colorectal cancer: A single-arm, open-label, multicentre phase 2 study. eClinicalMedicine 2023, 58, 101917. [Google Scholar] [CrossRef]
- Yarchoan, M.; Huang, C.; Zhu, Q.; Ferguson, A.K.; Durham, J.N.; Anders, R.A.; Thompson, E.D.; Rozich, N.S.; Thomas, D.L.; Nauroth, J.M.; et al. A phase 2 study of GVAX colon vaccine with cyclophosphamide and pembrolizumab in patients with mismatch repair proficient advanced colorectal cancer. Cancer Med. 2020, 9, 1485–1494. [Google Scholar] [CrossRef]
- Haag, G.M.; Springfeld, C.; Grün, B.; Apostolidis, L.; Zschäbitz, S.; Dietrich, M.; Berger, A.-K.; Weber, T.F.; Zoernig, I.; Schaaf, M.; et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer—The PICCASSO phase I trial. Eur. J. Cancer 2022, 167, 112–122. [Google Scholar] [CrossRef]
- Patel, M.R.; Falchook, G.S.; Hamada, K.; Makris, L.; Bendell, J.C. A phase 2 trial of trifluridine/tipiracil plus nivolumab in patients with heavily pretreated microsatellite-stable metastatic colorectal cancer. Cancer Med. 2021, 10, 1183–1190. [Google Scholar] [CrossRef]
- Kuang, C.; Park, Y.; Augustin, R.C.; Lin, Y.; Hartman, D.J.; Seigh, L.; Pai, R.K.; Sun, W.; Bahary, N.; Ohr, J.; et al. Pembrolizumab plus azacitidine in patients with chemotherapy refractory metastatic colorectal cancer: A single-arm phase 2 trial and correlative biomarker analysis. Clin. Epigenet. 2022, 14, 3. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Gorzo, A.; Galos, D.; Volovat, S.R.; Lungulescu, C.V.; Burz, C.; Sur, D. Landscape of Immunotherapy Options for Colorectal Cancer: Current Knowledge and Future Perspectives beyond Immune Checkpoint Blockade. Life 2022, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Zagorulya, M.; Spranger, S. Once upon a prime: DCs shape cancer immunity. Trends Cancer 2023, 9, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef]
- Kloor, M.; Michel, S.; Von Knebel Doeberitz, M. Immune evasion of microsatellite unstable colorectal cancers. Int. J. Cancer 2010, 127, 1001–1010. [Google Scholar] [CrossRef]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef]
- Rieth, J.; Subramanian, S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. Int. J. Mol. Sci. 2018, 19, 1340. [Google Scholar] [CrossRef]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated with the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef]
- Lichtenstern, C.R.; Ngu, R.K.; Shalapour, S.; Karin, M. Immunotherapy, Inflammation and Colorectal Cancer. Cells 2020, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Heregger, R.; Huemer, F.; Steiner, M.; Gonzalez-Martinez, A.; Greil, R.; Weiss, L. Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer. Cancers 2023, 15, 5090. [Google Scholar] [CrossRef]
- Wang, H.; Liu, B.; Wei, J. Beta2-microglobulin(B2M) in cancer immunotherapies: Biological function, resistance and remedy. Cancer Lett. 2021, 517, 96–104. [Google Scholar] [CrossRef]
- Lagos, G.G.; Izar, B.; Rizvi, N.A. Beyond Tumor PD-L1: Emerging Genomic Biomarkers for Checkpoint Inhibitor Immunotherapy. In American Society of Clinical Oncology Educational Book; Annual Meeting; American Society of Clinical Oncology: Alexandria, VA, USA, 2020; pp. e47–e57. [Google Scholar] [CrossRef]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef]
- Stelloo, E.; Versluis, M.A.; Nijman, H.W.; De Bruyn, M.; Plat, A.; Osse, E.M.; Van Dijk, R.H.; Nout, R.A.; Creutzberg, C.L.; De Bock, G.H.; et al. Microsatellite instability derived JAK1 frameshift mutations are associated with tumor immune evasion in endometrioid endometrial cancer. Oncotarget 2016, 7, 39885–39893. [Google Scholar] [CrossRef]
- Albacker, L.A.; Wu, J.; Smith, P.; Warmuth, M.; Stephens, P.J.; Zhu, P.; Yu, L.; Chmielecki, J. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS ONE 2017, 12, e0176181. [Google Scholar] [CrossRef]
- Nguyen, T.-T.; Ramsay, L.; Ahanfeshar-Adams, M.; Lajoie, M.; Schadendorf, D.; Alain, T.; Watson, I.R. Mutations in the IFNγ-JAK-STAT Pathway Causing Resistance to Immune Checkpoint Inhibitors in Melanoma Increase Sensitivity to Oncolytic Virus Treatment. Clin. Cancer Res. 2021, 27, 3432–3442. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Kyriakopoulou, G.; Papavassiliou, A.G.; Karamouzis, M.V. The Interplay of Autophagy and Tumor Microenvironment in Colorectal Cancer—Ways of Enhancing Immunotherapy Action. Cancers 2019, 11, 533. [Google Scholar] [CrossRef]
- Devenport, S.N.; Singhal, R.; Radyk, M.D.; Taranto, J.G.; Kerk, S.A.; Chen, B.; Goyert, J.W.; Jain, C.; Das, N.K.; Oravecz-Wilson, K.; et al. Colorectal cancer cells utilize autophagy to maintain mitochondrial metabolism for cell proliferation under nutrient stress. JCI Insight 2021, 6, e138835. [Google Scholar] [CrossRef]
- Zhang, P.; Cheng, S.; Sheng, X.; Dai, H.; He, K.; Du, Y. The role of autophagy in regulating metabolism in the tumor microenvironment. Genes Dis. 2023, 10, 447–456. [Google Scholar] [CrossRef]
- Manzoor, S.; Muhammad, J.S.; Maghazachi, A.A.; Hamid, Q. Autophagy: A Versatile Player in the Progression of Colorectal Cancer and Drug Resistance. Front. Oncol. 2022, 12, 924290. [Google Scholar] [CrossRef]
- Jin, C.; Wang, T.; Yang, Y.; Zhou, P.; Li, J.; Wu, W.; Lv, X.; Ma, G.; Wang, A. Rational targeting of autophagy in colorectal cancer therapy: From molecular interactions to pharmacological compounds. Environ. Res. 2023, 227, 115721. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, R.; PriyaDharshini, L.C.; Sakthivel, K.M.; Rasmi, R.R. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2022, 1868, 166400. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Sarry, J.-E.; Joffre, C. Autophagy is a major metabolic regulator involved in cancer therapy resistance. Cell Rep. 2021, 36, 109528. [Google Scholar] [CrossRef]
- Li, Y.; Li, W.; Hoffman, A.R.; Cui, J.; Hu, J.-F. The Nucleus/Mitochondria-Shuttling LncRNAs Function as New Epigenetic Regulators of Mitophagy in Cancer. Front. Cell Dev. Biol. 2021, 9, 699621. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.-D.; Yan, F.; Jing, Y.-Y.; Han, Z.-P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.-X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Claude-Taupin, A.; Jia, J.; Mudd, M.; Deretic, V. Autophagy’s secret life: Secretion instead of degradation. Essays Biochem. 2017, 61, 637–647. [Google Scholar] [CrossRef]
- Camuzard, O.; Santucci-Darmanin, S.; Carle, G.F.; Pierrefite-Carle, V. Autophagy in the crosstalk between tumor and microenvironment. Cancer Lett. 2020, 490, 143–153. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy Proteins Regulate the Secretory Component of Osteoclastic Bone Resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef]
- Cotzomi-Ortega, I.; Aguilar-Alonso, P.; Reyes-Leyva, J.; Maycotte, P. Autophagy and Its Role in Protein Secretion: Implications for Cancer Therapy. Mediators Inflamm. 2018, 2018, 4231591. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-L.; Yuan, L.; Tang, Y.-C.; Xu, Z.-Y.; Xu, H.-D.; Cheng, X.-D.; Qin, J.-J. The Role of Autophagy in Gastric Cancer Chemoresistance: Friend or Foe? Front. Cell Dev. Biol. 2020, 8, 621428. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Farhana, A.; Alsrhani, A.; Khan, Y.S.; Rasheed, Z. Cancer Bioenergetics and Tumor Microenvironments—Enhancing Chemotherapeutics and Targeting Resistant Niches through Nanosystems. Cancers 2023, 15, 3836. [Google Scholar] [CrossRef]
- Jiao, L.; Zhang, H.-L.; Li, D.-D.; Yang, K.-L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.-Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-Dependent Production of Secreted Factors Facilitates Oncogenic RAS-Driven Invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth and Metastasis via Oxidative Stress, Mitophagy, and Aerobic Glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Noma, T.; Makino, T.; Ohshima, K.; Sugimura, K.; Miyata, H.; Honma, K.; Yamashita, K.; Saito, T.; Tanaka, K.; Yamamoto, K.; et al. Immunoscore Signatures in Surgical Specimens and Tumor-Infiltrating Lymphocytes in Pretreatment Biopsy Predict Treatment Efficacy and Survival in Esophageal Cancer. Ann. Surg. 2023, 277, e528–e537. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC Class II Processing of a Viral Nuclear Antigen After Autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Münz, C. Antigen processing via autophagy—Not only for MHC class II presentation anymore? Curr. Opin. Immunol. 2010, 22, 89–93. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Loi, M.; Müller, A.; Steinbach, K.; Niven, J.; Barreira Da Silva, R.; Paul, P.; Ligeon, L.-A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti-viral CD8 + T Cell Responses. Cell Rep. 2016, 15, 1076–1087. [Google Scholar] [CrossRef]
- Alissafi, T.; Hatzioannou, A.; Mintzas, K.; Barouni, R.M.; Banos, A.; Sormendi, S.; Polyzos, A.; Xilouri, M.; Wielockx, B.; Gogas, H.; et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Investig. 2018, 128, 3840–3852. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Parekh, V.V.; Pabbisetty, S.K.; Wu, L.; Sebzda, E.; Martinez, J.; Zhang, J.; Van Kaer, L. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8α+ dendritic cells. Proc. Natl. Acad. Sci. USA 2017, 114, E6371–E6380. [Google Scholar] [CrossRef]
- Yang, G.; Song, W.; Postoak, J.L.; Chen, J.; Martinez, J.; Zhang, J.; Wu, L.; Van Kaer, L. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function: PIK3C3/VPS34 in T cell metabolism and function. Autophagy 2021, 17, 1193–1204. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Theoharis, S.; Saetta, A.A.; Chatziandreou, I.; Kyriakopoulou, G.; Giannopoulou, I.; Michelli, M.; Schizas, D.; Papavassiliou, A.G.; et al. Autophagy-related Proteins as a Prognostic Factor of Patients with Colorectal Cancer. Am. J. Clin. Oncol. 2019, 42, 767–776. [Google Scholar] [CrossRef]
- Yang, Z.; Ghoorun, R.A.; Fan, X.; Wu, P.; Bai, Y.; Li, J.; Chen, H.; Wang, L.; Wang, J. High expression of Beclin-1 predicts favorable prognosis for patients with colorectal cancer. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 98–106. [Google Scholar] [CrossRef]
- Zaanan, A.; Park, J.M.; Tougeron, D.; Huang, S.; Wu, T.-T.; Foster, N.R.; Sinicrope, F.A. Association of beclin 1 expression with response to neoadjuvant chemoradiation therapy in patients with locally advanced rectal carcinoma: Beclin 1 Predicts Response to Chemoradiation. Int. J. Cancer 2015, 137, 1498–1502. [Google Scholar] [CrossRef]
- Yang, K.; Park, C.G.; Cheong, C.; Bulgheresi, S.; Zhang, S.; Zhang, P.; He, Y.; Jiang, L.; Huang, H.; Ding, H.; et al. Host Langerin (CD207) is a receptor for Yersinia pestis phagocytosis and promotes dissemination. Immunol. Cell Biol. 2015, 93, 815–824. [Google Scholar] [CrossRef]
- Burada, F. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271. [Google Scholar] [CrossRef]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- Mahgoub, E.; Taneera, J.; Sulaiman, N.; Saber-Ayad, M. The role of autophagy in colorectal cancer: Impact on pathogenesis and implications in therapy. Front. Med. 2022, 9, 959348. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Mele, L.; Del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Ariosa, A.R.; Lahiri, V.; Lei, Y.; Yang, Y.; Yin, Z.; Zhang, Z.; Klionsky, D.J. A perspective on the role of autophagy in cancer. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2021, 1867, 166262. [Google Scholar] [CrossRef]
- Dong, Y.; Wu, Y.; Zhao, G.-L.; Ye, Z.-Y.; Xing, C.-G.; Yang, X.-D. Inhibition of autophagy by 3-MA promotes hypoxia-induced apoptosis in human colorectal cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1047–1054. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, M.-M.; Hou, J.-G. Molecular and cellular pathways in colorectal cancer: Apoptosis, autophagy and inflammation as key players. Scand. J. Gastroenterol. 2022, 57, 1279–1290. [Google Scholar] [CrossRef]
- Rangel, M.; Kong, J.; Bhatt, V.; Khayati, K.; Guo, J.Y. Autophagy and tumorigenesis. FEBS J. 2022, 289, 7177–7198. [Google Scholar] [CrossRef]
- Pandey, A.; Yadav, P.; Shukla, S. Unfolding the role of autophagy in the cancer metabolism. Biochem. Biophys. Rep. 2021, 28, 101158. [Google Scholar] [CrossRef]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A challengeable paradox in cancer treatment. Cancer Med. 2023, 12, 11542–11569. [Google Scholar] [CrossRef]
- Vasilevskaya, I.A.; Selvakumaran, M.; Roberts, D.; O’Dwyer, P.J. JNK1 Inhibition Attenuates Hypoxia-Induced Autophagy and Sensitizes to Chemotherapy. Mol. Cancer Res. 2016, 14, 753–763. [Google Scholar] [CrossRef]
- Wen, Y.-A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Scott, A.J.; Arcaroli, J.J.; Bagby, S.M.; Yahn, R.; Huber, K.M.; Serkova, N.J.; Nguyen, A.; Kim, J.; Thorburn, A.; Vogel, J.; et al. Cabozantinib Exhibits Potent Antitumor Activity in Colorectal Cancer Patient-Derived Tumor Xenograft Models via Autophagy and Signaling Mechanisms. Mol. Cancer Ther. 2018, 17, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Zhang, W.; Yuan, L.; Tan, J.; Chen, Z. HIF-1a regulates hypoxia-induced autophagy via translocation of ANKRD37 in colon cancer. Exp. Cell Res. 2020, 395, 112175. [Google Scholar] [CrossRef]
- Belisario, D.C.; Kopecka, J.; Pasino, M.; Akman, M.; De Smaele, E.; Donadelli, M.; Riganti, C. Hypoxia Dictates Metabolic Rewiring of Tumors: Implications for Chemoresistance. Cells 2020, 9, 2598. [Google Scholar] [CrossRef]
- Metur, S.P.; Klionsky, D.J. Adaptive immunity at the crossroads of autophagy and metabolism. Cell. Mol. Immunol. 2021, 18, 1096–1105. [Google Scholar] [CrossRef]
- Hubbard, V.M.; Valdor, R.; Patel, B.; Singh, R.; Cuervo, A.M.; Macian, F. Macroautophagy Regulates Energy Metabolism during Effector T Cell Activation. J. Immunol. 2010, 185, 7349–7357. [Google Scholar] [CrossRef]
- Virgin, H.W.; Levine, B. Autophagy genes in immunity. Nat. Immunol. 2009, 10, 461–470. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Li, C.; Capan, E.; Zhao, Y.; Zhao, J.; Stolz, D.; Watkins, S.C.; Jin, S.; Lu, B. Autophagy Is Induced in CD4+ T Cells and Important for the Growth Factor-Withdrawal Cell Death. J. Immunol. 2006, 177, 5163–5168. [Google Scholar] [CrossRef]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.-W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007, 204, 25–31. [Google Scholar] [CrossRef]
- Yu, Q.; Ding, J.; Li, S.; Li, Y. Autophagy in cancer immunotherapy: Perspective on immune evasion and cell death interactions. Cancer Lett. 2024, 590, 216856. [Google Scholar] [CrossRef]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef]
- Wen, Z.-F.; Liu, H.; Gao, R.; Zhou, M.; Ma, J.; Zhang, Y.; Zhao, J.; Chen, Y.; Zhang, T.; Huang, F.; et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer 2018, 6, 151. [Google Scholar] [CrossRef]
- Roman, V.; Mihaila, M.; Radu, N.; Marineata, S.; Diaconu, C.C.; Bostan, M. Cell Culture Model Evolution and Its Impact on Improving Therapy Efficiency in Lung Cancer. Cancers 2023, 15, 4996. [Google Scholar] [CrossRef]
- Fessas, P.; Lee, H.; Ikemizu, S.; Janowitz, T. A molecular and preclinical comparison of the PD-1–targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin. Oncol. 2017, 44, 136–140. [Google Scholar] [CrossRef]
- Barham, W.; Hsu, M.; Liu, X.; Harrington, S.M.; Hirdler, J.B.; Gicobi, J.K.; Zhu, X.; Zeng, H.; Pavelko, K.D.; Yan, Y.; et al. A Novel Humanized PD-1/PD-L1 Mouse Model Permits Direct Comparison of Antitumor Immunity Generated by Food and Drug Administration–Approved PD-1 and PD-L1 Inhibitors. ImmunoHorizons 2023, 7, 125–139. [Google Scholar] [CrossRef]
- Craig, A.W.; Frieboes, H.B.; Videira, P.A. Advancing cancer immunotherapy: From innovative preclinical models to clinical insights. Sci. Rep. 2024, 14, 1205. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472. [Google Scholar] [CrossRef]
- Tenorio-Pedraza, J.M.; Lippert, J.; Burghaus, R.; Scheerans, C. Meta-analysis of preclinical measures of efficacy in immune checkpoint blockade therapies and comparison to clinical efficacy estimates. Transl. Med. Commun. 2023, 8, 17. [Google Scholar] [CrossRef]
- Durinikova, E.; Buzo, K.; Arena, S. Preclinical models as patients’ avatars for precision medicine in colorectal cancer: Past and future challenges. J. Exp. Clin. Cancer Res. 2021, 40, 185. [Google Scholar] [CrossRef]
- Donahue, R.N.; Lepone, L.M.; Grenga, I.; Jochems, C.; Fantini, M.; Madan, R.A.; Heery, C.R.; Gulley, J.L.; Schlom, J. Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti-PD-L1 monoclonal antibody. J. Immunother. Cancer 2017, 5, 20. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.-S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Bareham, B.; Georgakopoulos, N.; Matas-Céspedes, A.; Curran, M.; Saeb-Parsy, K. Modeling human tumor-immune environments in vivo for the preclinical assessment of immunotherapies. Cancer Immunol. Immunother. 2021, 70, 2737–2750. [Google Scholar] [CrossRef]
- Shang, P.; Yu, L.; Cao, S.; Guo, C.; Zhang, W. An improved cell line-derived xenograft humanized mouse model for evaluation of PD-1/PD-L1 blocker BMS202-induced immune responses in colorectal cancer. Acta Biochim. Biophys. Sin. 2022, 54, 1497–1506. [Google Scholar] [CrossRef]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef]
- Kabeya, Y. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Muñoz-Galdeano, T.; Reigada, D.; Del Águila, Á.; Velez, I.; Caballero-López, M.J.; Maza, R.M.; Nieto-Díaz, M. Cell Specific Changes of Autophagy in a Mouse Model of Contusive Spinal Cord Injury. Front. Cell. Neurosci. 2018, 12, 164. [Google Scholar] [CrossRef]
- Mainz, L.; Rosenfeldt, M.T. Autophagy and cancer—Insights from mouse models. FEBS J. 2018, 285, 792–808. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Hashemi, M.; Mohandesi Khosroshahi, E.; Tanha, M.; Khoushab, S.; Bizhanpour, A.; Azizi, F.; Mohammadzadeh, M.; Matinahmadi, A.; Khazaei Koohpar, Z.; Asadi, S.; et al. Targeting autophagy can synergize the efficacy of immune checkpoint inhibitors against therapeutic resistance: New promising strategy to reinvigorate cancer therapy. Heliyon 2024, 10, e37376. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.-P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L.; et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 2019, 3, 306–317. [Google Scholar] [CrossRef]
- Li, Y.; Hahn, T.; Garrison, K.; Cui, Z.-H.; Thorburn, A.; Thorburn, J.; Hu, H.-M.; Akporiaye, E.T. The Vitamin E Analogue α-TEA Stimulates Tumor Autophagy and Enhances Antigen Cross-Presentation. Cancer Res. 2012, 72, 3535–3545. [Google Scholar] [CrossRef]
- Hahn, T.; Akporiaye, E.T. α-TEA as a stimulator of tumor autophagy and enhancer of antigen cross-presentation. Autophagy 2013, 9, 429–431. [Google Scholar] [CrossRef]
- Diem, S.; Hasan Ali, O.; Ackermann, C.J.; Bomze, D.; Koelzer, V.H.; Jochum, W.; Speiser, D.E.; Mertz, K.D.; Flatz, L. Tumor infiltrating lymphocytes in lymph node metastases of stage III melanoma correspond to response and survival in nine patients treated with ipilimumab at the time of stage IV disease. Cancer Immunol. Immunother. 2018, 67, 39–45. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201.e10. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, L.; Liu, J.; Chen, B.; Shi, H.; Chen, H.; Qi, H.; Wu, Z.; Mao, X.; Wang, X.; et al. Inhibition of autophagy-related protein 7 enhances anti-tumor immune response and improves efficacy of immune checkpoint blockade in microsatellite instability colorectal cancer. J. Exp. Clin. Cancer Res. 2024, 43, 114. [Google Scholar] [CrossRef]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. OncoImmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Robainas, M.; Otano, R.; Bueno, S.; Ait-Oudhia, S. Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. OncoTargets Ther. 2017, 10, 1803–1807. [Google Scholar] [CrossRef]
- Saleh, T.; Cuttino, L.; Gewirtz, D.A. Autophagy is not uniformly cytoprotective: A personalized medicine approach for autophagy inhibition as a therapeutic strategy in non-small cell lung cancer. Biochim. Biophys. Acta BBA Gen. Subj. 2016, 1860, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.-D.; Blanchet, B.; Periou, B.; Authier, F.-J.; Mograbi, B.; Gherardi, R.K.; Crépeaux, G. Long Term Pharmacological Perturbation of Autophagy in Mice: Are HCQ Injections a Relevant Choice? Biomedicines 2020, 8, 47. [Google Scholar] [CrossRef]
- Wabitsch, S.; McVey, J.C.; Ma, C.; Ruf, B.; Kamenyeva, O.; McCallen, J.D.; Diggs, L.P.; Heinrich, B.; Greten, T.F. Hydroxychloroquine can impair tumor response to anti-PD1 in subcutaneous mouse models. iScience 2021, 24, 101990. [Google Scholar] [CrossRef]
- Krueger, J.; Santinon, F.; Kazanova, A.; Issa, M.E.; Larrivee, B.; Kremer, R.; Milhalcioiu, C.; Rudd, C.E. Hydroxychloroquine (HCQ) decreases the benefit of anti-PD-1 immune checkpoint blockade in tumor immunotherapy. PLoS ONE 2021, 16, e0251731. [Google Scholar] [CrossRef]
- Sharma, G.; Ojha, R.; Noguera-Ortega, E.; Rebecca, V.W.; Attanasio, J.; Liu, S.; Piao, S.; Lee, J.J.; Nicastri, M.C.; Harper, S.L.; et al. PPT1 inhibition enhances the antitumor activity of anti–PD-1 antibody in melanoma. JCI Insight 2020, 5, e133225. [Google Scholar] [CrossRef]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef]
- Li, X.; Zhu, F.; Jiang, J.; Sun, C.; Zhong, Q.; Shen, M.; Wang, X.; Tian, R.; Shi, C.; Xu, M.; et al. Simultaneous inhibition of the ubiquitin-proteasome system and autophagy enhances apoptosis induced by ER stress aggravators in human pancreatic cancer cells. Autophagy 2016, 12, 1521–1537. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The Challenge of Developing Autophagy Inhibition as a Therapeutic Strategy. Cancer Res. 2016, 76, 5610–5614. [Google Scholar] [CrossRef]
- Kong, J.; Xu, S.; Zhang, P.; Zhao, Y. CXCL1 promotes immune escape in colorectal cancer by autophagy-mediated MHC-I degradation. Hum. Immunol. 2023, 84, 110716. [Google Scholar] [CrossRef]
- Ishimwe, N.; Wei, P.; Wang, M.; Zhang, H.; Wang, L.; Jing, M.; Wen, L.; Zhang, Y. Autophagy Impairment through Lysosome Dysfunction by Brucine Induces Immunogenic Cell Death (ICD). Am. J. Chin. Med. 2020, 48, 1915–1940. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Qian, J.; Wei, G.; Ding, R.; Hu, C.; Fang, D.; Jiang, Z.; Bi, L.; Song, J.; et al. FuFangChangTai Decoction Activates Macrophages via Inducing Autophagy. Evid. Based Complement. Alternat. Med. 2019, 2019, 5657035. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Guo, H.; Zhang, J.; Zheng, M.; Zhang, W.; Wang, L.; Du, Q.; Zhou, C.; Xu, Y.; Wu, H.; et al. Zosuquidar Promotes Antitumor Immunity by Inducing Autophagic Degradation of PD-L1. Adv. Sci. 2024, 11, 202400340. [Google Scholar] [CrossRef]
- Zhou, X.; Fu, D.; Yang, H.; Le, C.; Lu, Y.; Wei, J.; Tang, Y.; Zhang, J.; Yuan, Y.; Ding, K.; et al. Rigosertib promotes anti-tumor immunity via autophagic degradation of PD-L1 in colorectal cancer cells. Cancer Lett. 2023, 577, 216422. [Google Scholar] [CrossRef]
- Maisonneuve, C.; Tsang, D.K.L.; Foerster, E.G.; Robert, L.M.; Mukherjee, T.; Prescott, D.; Tattoli, I.; Lemire, P.; Winer, D.A.; Winer, S.; et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep. 2021, 34, 108677. [Google Scholar] [CrossRef]
- Morschhauser, F.; Machiels, J.-P.; Salles, G.; Rottey, S.; Rule, S.A.J.; Cunningham, D.; Peyrade, F.; Fruchart, C.; Arkenau, H.-T.; Genvresse, I.; et al. On-Target Pharmacodynamic Activity of the PI3K Inhibitor Copanlisib in Paired Biopsies from Patients with Malignant Lymphoma and Advanced Solid Tumors. Mol. Cancer Ther. 2020, 19, 468–478. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Arakelian, T.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti–PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
ALKhemeiri, N.; Eljack, S.; Saber-Ayad, M.M. Perspectives of Targeting Autophagy as an Adjuvant to Anti-PD-1/PD-L1 Therapy for Colorectal Cancer Treatment. Cells 2025, 14, 745. https://doi.org/10.3390/cells14100745
ALKhemeiri N, Eljack S, Saber-Ayad MM. Perspectives of Targeting Autophagy as an Adjuvant to Anti-PD-1/PD-L1 Therapy for Colorectal Cancer Treatment. Cells. 2025; 14(10):745. https://doi.org/10.3390/cells14100745
Chicago/Turabian StyleALKhemeiri, Nasrah, Sahar Eljack, and Maha Mohamed Saber-Ayad. 2025. "Perspectives of Targeting Autophagy as an Adjuvant to Anti-PD-1/PD-L1 Therapy for Colorectal Cancer Treatment" Cells 14, no. 10: 745. https://doi.org/10.3390/cells14100745
APA StyleALKhemeiri, N., Eljack, S., & Saber-Ayad, M. M. (2025). Perspectives of Targeting Autophagy as an Adjuvant to Anti-PD-1/PD-L1 Therapy for Colorectal Cancer Treatment. Cells, 14(10), 745. https://doi.org/10.3390/cells14100745