
Unraveling Glypican-3: From Structural to Pathophysiological Roles and Mechanisms—An Integrative Perspective


Abstract
1. Introduction
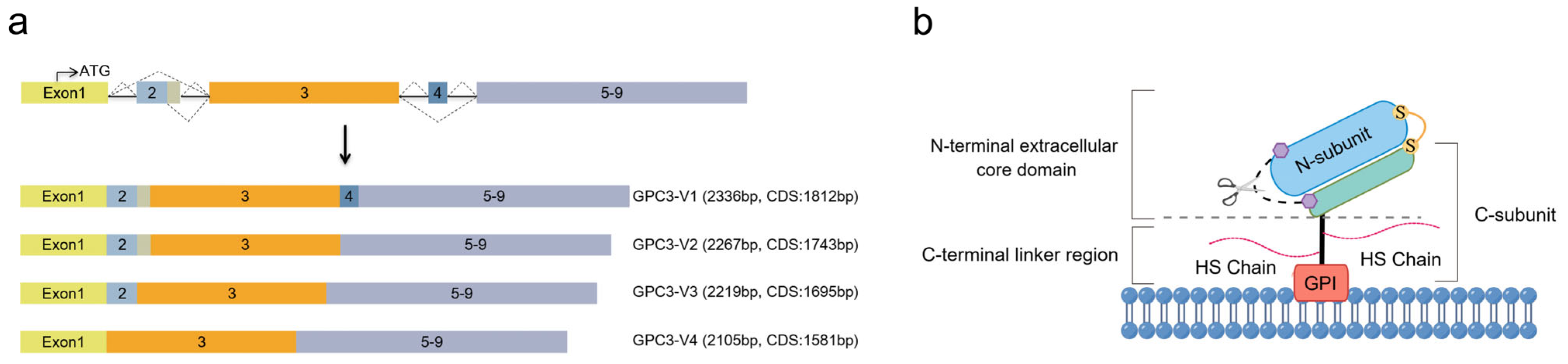
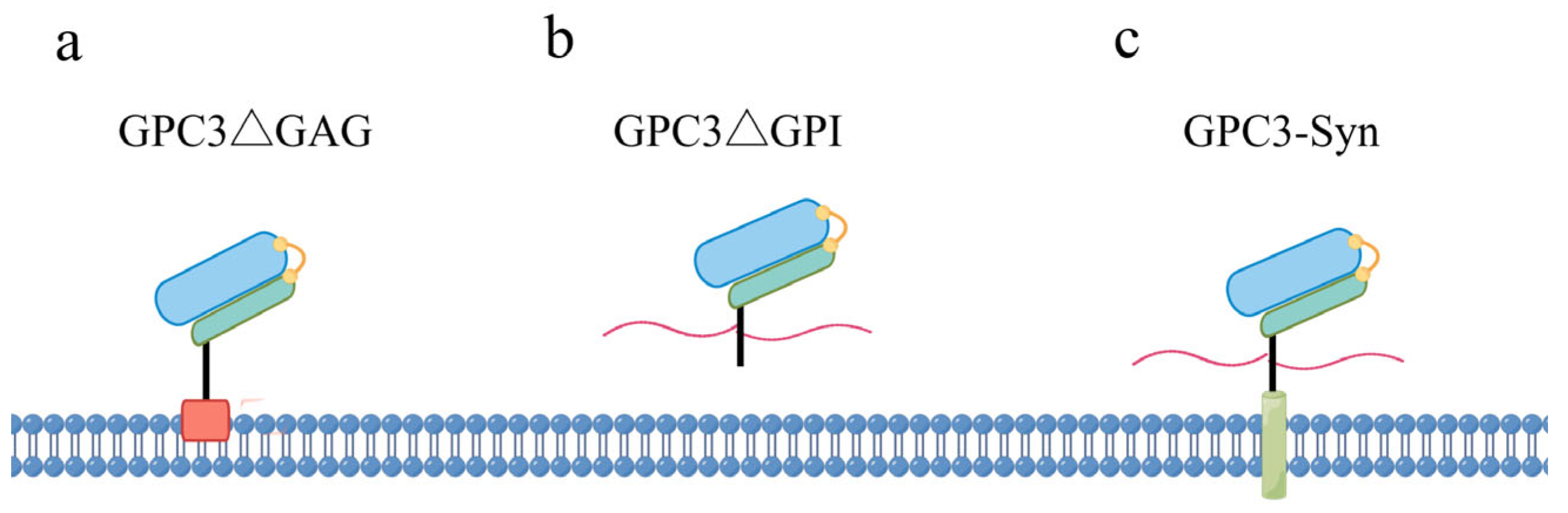
2. The Structural Features of GPC3
3. Physiological Functions and Mechanisms of GPC3
3.1. Physiological Functions of GPC3
3.2. Mechanisms Underlying GPC3’s Physiological Function
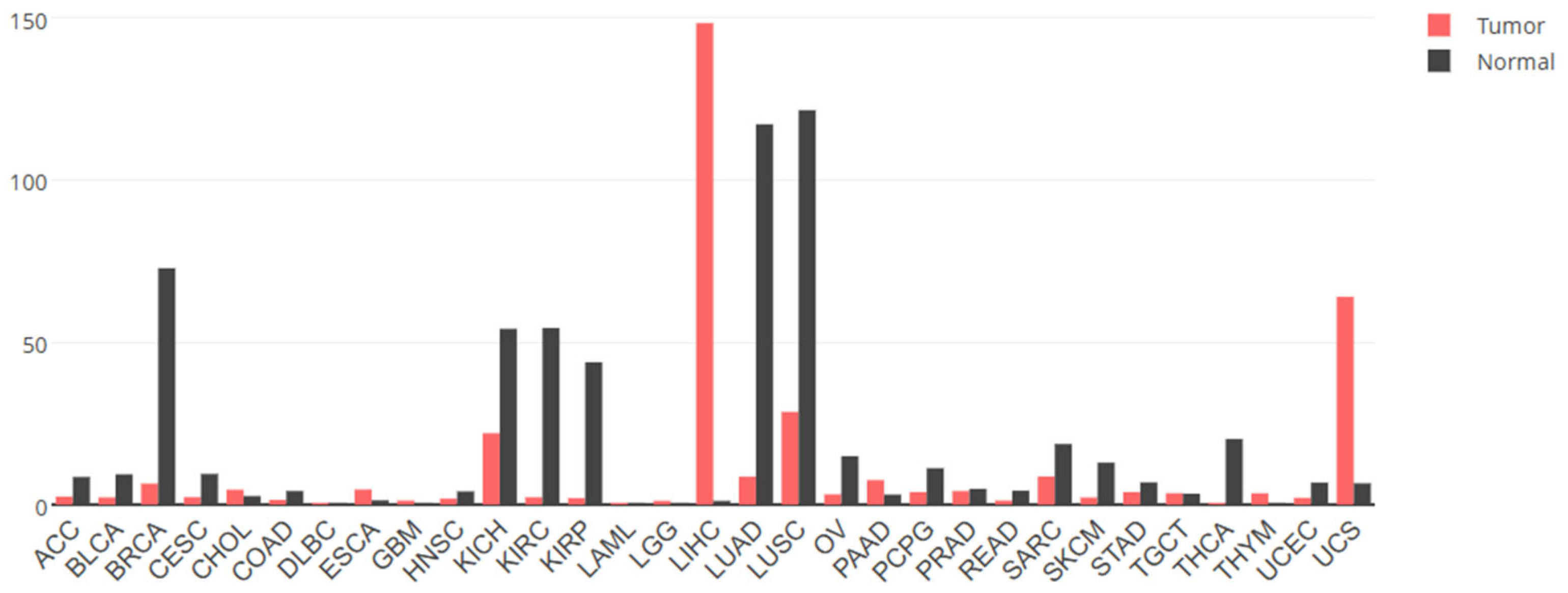
4. GPC3 in Cancer
4.1. GPC3 as an Oncogene in Cancers
4.1.1. Oncogenic Roles of GPC3
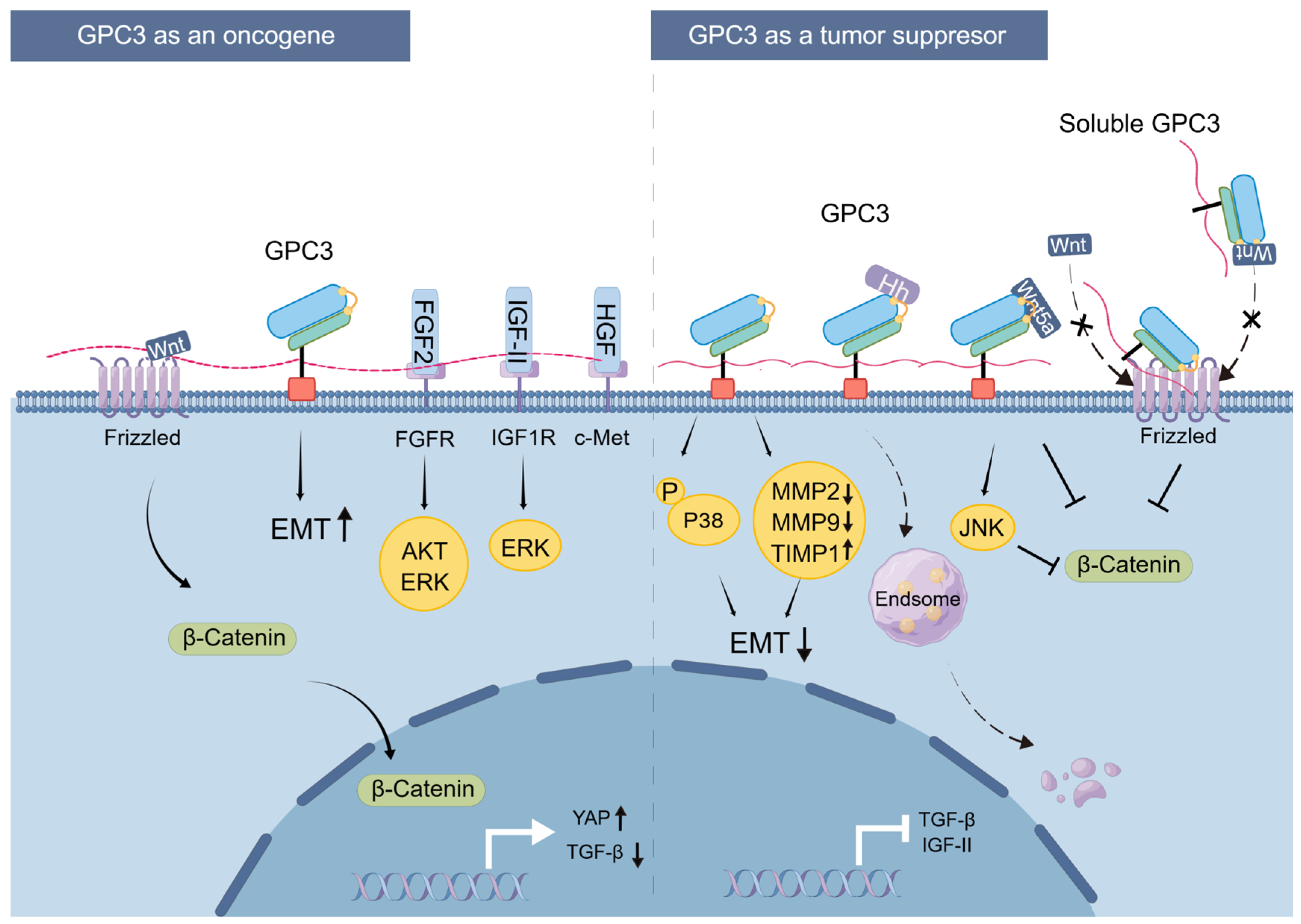
4.1.2. Pro-Oncogenic Mechanism of GPC3
4.2. GPC3 as a Tumor Suppressor in Cancers
4.2.1. Anti-Oncogenic Role of GPC3
4.2.2. Mechanism of Cancer Inhibition by GPC3
5. Relationship Between the Structural Features of GPC3 and Its Functions
5.1. GPC3’s Structural Features and Its Physiological Functions
5.2. GPC3’s Structural Features and Its Oncogenic Function
5.3. GPC3’s Structural Features and Its Anti-Oncogenic Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | HS | Convertase | Anchored | Soluble | N-Glycan | ||
|---|---|---|---|---|---|---|---|
| Effectors | |||||||
| Physiological function | (anti-) Hh | No [46], Yes [111] | Yes [112] | Yes [46] | No [46] | - | |
| (pro-) Unc5D | - | No [50] | - | - | Yes [50] | ||
| pro-oncogenic function | (pro-) canonical Wnt | No [82], Yes [114] | No [12] | Yes [117] | No [117] | - | |
| (pro-) IGF-II | - | Yes [85] | - | - | - | ||
| (pro-) FGF2 | Yes [97,116] | - | - | - | - | ||
| (pro-) HGF | Yes [98] | - | - | - | - | ||
| anti-oncogenic Functions | (pro-) non-canonical Wnt | No [11] | Yes [13] | Yes [11] | - | - | |
| (anti-) IGF-II | - | - | - | Yes [63] | - | ||
| (anti-) CD26+ | No [118] | - | - | Yes [118] | - | ||
| (anti-) canonical Wnt | - | - | - | Yes [117] | - | ||
6. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | Adrenocortical cancer |
| AFP | Alpha-fetoprotein |
| BLCA | Bladder cancer |
| BM | Bone marrow |
| BMP | Bone morphogenetic protein |
| BRCA | Breast cancer |
| BWS | Beckwith–Wiedemann syndrome |
| CAF | Cancer-associated fibroblast |
| CCC | Clear cell carcinoma |
| CESC | Cervical cancer |
| CHOL | Bile duct cancer |
| COAD | Colon cancer |
| DLBC | Large B-cell lymphoma |
| EMT | Epithelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| ESCA | Esophageal cancer |
| ESCC | Esophageal squamous cell carcinoma |
| FZD | Frizzled |
| GBM | Glioblastoma |
| GC | Gastric cancer |
| GPC | Glypican |
| GPC3 | Glypican3 |
| GPI | Glycosylphosphatidylinositol |
| GLUT4 | Glucose transporter4 |
| HCC | Hepatocellular carcinoma |
| hESC | Human embryonic stem cell |
| HGF | Hepatocyte growth factor |
| Hh | Hedgehog |
| Hhex | Hematopoietically expressed homeobox |
| HNSC | Head and neck cancer |
| HS | Heparan sulfate |
| HSPC | Hematopoietic stem/progenitor cells |
| HSPG | Heparan sulfate proteoglycan |
| IGF-II | Insulin-like growth factor II |
| IGF1R | IGF receptor-1 |
| IRS-1 | Insulin receptor substrate-1 |
| KGF | Keratinocyte growth factor |
| KICH | Kidney chromophobe |
| KIRC | Kidney clear cell carcinoma |
| KIRP | Kidney papillary cell carcinoma |
| LAML | Acute myeloid leukemia |
| LGG | Lower-grade glioma |
| LIHC | Liver hepatocellular cancer |
| LRP1 | Low-density lipoprotein receptor-related protein-1 |
| LUAD | Lung adenocarcinoma |
| LUNG | Lung cancer |
| LUSC | Lung squamous cell carcinoma |
| MAPK | Mitogen-activated protein kinase |
| MEF | Mouse embryonic fibroblast |
| MMP | Matrix metalloproteinase |
| OCCA | Ovarian clear cell carcinoma |
| OV | Ovarian cancer |
| PAAD | Pancreatic cancer |
| PCPG | Pheochromocytoma and paraganglioma |
| READ | Rectal cancer |
| PRAD | Prostate cancer |
| Ptc | Patched |
| SARC | Sarcoma |
| SGBS | Simpson–Golabi–Behmel syndrome |
| Shh | Sonic Hedgehog |
| SKCM | Skin cutaneous melanoma |
| STAD | Stomach cancer |
| SULF2 | Sulfate esterase 2 |
| TGCT | Testicular cancer |
| TGF-β | Transforming growth factor-β |
| THCA | Thyroid cancer |
| THYM | Thymoma |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| TIMP-1 | Tissue inhibitors of metalloproteinase 1 |
| UCEC | Endometrioid cancer |
| UCS | Uterine carcinosarcoma |
| Unc5D | Uncoordinated-5 receptor D |
| V2 | Variant 2 |
| YAP | Yes-associated protein |
References
- Filmus, J.; Church, J.G.; Buick, R.N. Isolation of a cDNA corresponding to a developmentally regulated transcript in rat intestine. Mol. Cell. Biol. 1988, 8, 4243–4249. [Google Scholar] [PubMed]
- Pilia, G.; HughesBenzie, R.M.; MacKenzie, A.; Baybayan, P.; Chen, E.Y.; Huber, R.; Neri, G.; Cao, A.; Forabosco, A.; Schlessinger, D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 1996, 12, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Sonoda, G.; Hamid, J.; Li, M.; Filmus, J.; Buick, R.N.; Testa, J.R. Mapping of the Simpson-Golabi-Behmel overgrowth syndrome gene (GPC3) to Chromosome X in human and rat by fluorescence in situ hybridization. Mamm. Genome 1997, 8, 72. [Google Scholar] [CrossRef]
- Lage, H.; Dietel, M. Cloning and characterization of human cDNAs encoding a protein with high homology to rat intestinal development protein OCI-5. Gene 1997, 188, 151–156. [Google Scholar] [CrossRef]
- Montalbano, M.; Georgiadis, J.; Masterson, A.L.; McGuire, J.T.; Prajapati, J.; Shirafkan, A.; Rastellini, C.; Cicalese, L. Biology and function of glypican-3 as a candidate for early cancerous transformation of hepatocytes in hepatocellular carcinoma (Review). Oncol. Rep. 2017, 37, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Tojjari, A.; Hafez, A.H.; Saeed, A.; Singh, M.; Saeed, A. Exploring Glypican-3 as a Molecular Target in Hepatocellular Carcinoma: Perspectives on Diagnosis and Precision Immunotherapy Strategies. Front. Biosci. 2024, 29, 268. [Google Scholar] [CrossRef]
- Tehrani, H.A.; Zangi, M.; Fathi, M.; Vakili, K.; Hassan, M.; Rismani, E.; Hossein-Khannazer, N.; Vosough, M. GPC-3 in hepatocellular carcinoma; A novel biomarker and molecular target. Exp. Cell Res. 2025, 444, 114391. [Google Scholar] [CrossRef]
- Couzinet, A.; Suzuki, T.; Nakatsura, T. Progress and challenges in glypican-3 targeting for hepatocellular carcinoma therapy. Expert Opin. Ther. Targets 2024, 28, 895–909. [Google Scholar] [CrossRef]
- Rismani, E.; Hossein-Khannazer, N.; Hassan, M.; Shams, E.; Najimi, M.; Vosough, M. Targeting glypican 3 by immunotoxins: The promise of immunotherapy in hepatocellular carcinoma. Expert Opin. Ther. Targets 2025, 29, 59–73. [Google Scholar] [CrossRef]
- Gonzalez, A.D.; Kaya, M.; Shi, W.; Song, H.; Testa, J.R.; Penn, L.Z.; Filmus, J. OCI-5/GPC3, a Glypican Encoded by a Gene That Is Mutated in the Simpson-Golabi-Behmel Overgrowth Syndrome, Induces Apoptosis in a Cell Line–specific Manner. J. Cell Biol. 1998, 141, 1407–1414. [Google Scholar] [CrossRef]
- Capurro, M.I.; Shi, W.; Sandal, S.; Filmus, J. Processing by convertases is not required for glypican-3-induced stimulation of hepatocellular carcinoma growth. J. Biol. Chem. 2005, 280, 41201–41206. [Google Scholar] [CrossRef]
- De Cat, B.; Muyldermans, S.Y.; Coomans, C.; Degeest, G.; Vanderschueren, B.; Creemers, J.; Biemar, F.; Peers, B.; David, G. Processing by proprotein convertases is required for glypican-3 modulation of cell survival, Wnt signaling, and gastrulation movements. J. Cell Biol. 2003, 163, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Stipp, C.S.; Litwack, E.D.; Lander, A.D. Cerebroglycan: An integral membrane heparan sulfate proteoglycan that is unique to the developing nervous system and expressed specifically during neuronal differentiation. J. Cell Biol. 1994, 124, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Traister, A.; Shi, W.; Filmus, J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem. J. 2008, 410, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a surprise. J. Clin. Investig. 2001, 108, 497–501. [Google Scholar] [CrossRef]
- Lin, A.E. Further delineation of the Simpson-Golabi-Behmel (SGB) syndrome. Am. J. Med. Genet. 1993, 46, 606–607. [Google Scholar] [CrossRef]
- Behmel, A.; Plochl, E.; Rosenkranz, W. A new X-linked dysplasia gigantism syndrome: Identical with the Simpson dysplasia syndrome? Hum. Genet. 1984, 67, 409–413. [Google Scholar] [CrossRef]
- Golabi, M.; Rosen, L. A new X-linked mental retardation-overgrowth syndrome. Am. J. Med. Genet. 1984, 17, 345–358. [Google Scholar] [CrossRef]
- Garganta, C.L.; Bodurtha, J.N. Report of another family with Simpson-Golabi-Behmel syndrome and a review of the literature. Am. J. Med. Genet. 1992, 44, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, S.; Ireland, M.; Obrien, O.; ClaytonSmith, J.; Hurst, J.A.; Mann, J.; Cole, T.; Sampson, J.; Slaney, S.; Schlessinger, D.; et al. Large scale deletions in the GPC3 gene may account for a minority of cases of Simpson-Golabi-Behmel syndrome. J. Med. Genet. 1997, 34, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Veugelers, M.; De Cat, B.; Muyldermans, S.Y.; Reekmans, G.; Delande, N.; Frints, S.; Legius, E.; Fryns, J.P.; Schrander-Stumpel, C.; Weidle, B.; et al. Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: Identification of loss-of-function mutations in the GPC3 gene. Hum. Mol. Genet. 2000, 9, 1321–1328. [Google Scholar] [CrossRef]
- HughesBenzie, R.M.; Pilia, G.; Xuan, J.Y.; Hunter, A.G.W.; Chen, E.; Golabi, M.; Hurst, J.A.; Kobori, J.; Marymee, K.; Pagon, R.A.; et al. Simpson-Golabi-Behmel syndrome: Genotype/phenotype analysis of 18 affected males from 7 unrelated families. Am. J. Med. Genet. 1996, 66, 227–234. [Google Scholar] [CrossRef]
- Xuan, J.Y.; Hughes-Benzie, R.M.; MacKenzie, A.E. A small interstitial deletion in the GPC3 gene causes Simpson-Golabi-Behmel syndrome in a Dutch-Canadian family. J. Med. Genet. 1999, 36, 57–58. [Google Scholar] [CrossRef]
- Cano-Gauci, D.F.; Song, H.H.; Yang, H.; McKerlie, C.; Choo, B.; Shi, W.; Pullano, R.; Piscione, T.D.; Grisaru, S.; Soon, S.; et al. Glypican-3–Deficient Mice Exhibit Developmental Overgrowth and Some of the Abnormalities Typical of Simpson-Golabi-Behmel Syndrome. J. Cell Biol. 1999, 146, 255–264. [Google Scholar] [CrossRef]
- Paine-Saunders, S.; Viviano, B.L.; Zupicich, J.; Skarnes, W.C.; Saunders, S. glypican-3 Controls Cellular Responses to Bmp4 in Limb Patterning and Skeletal Development. Dev. Biol. 2000, 225, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Viviano, B.L.; Silverstein, L.; Pflederer, C.; Paine-Saunders, S.; Mills, K.; Saunders, S. Altered hematopoiesis in glypican-3-deficient mice results in decreased osteoclast differentiation and a delay in endochondral ossification. Dev. Biol. 2005, 282, 152–162. [Google Scholar] [CrossRef]
- Ng, A.; Wong, M.; Viviano, B.; Erlich, J.M.; Alba, G.; Pflederer, C.; Jay, P.Y.; Saunders, S. Loss of glypican-3 function causes growth factor-dependent defects in cardiac and coronary vascular development. Dev. Biol. 2009, 335, 208–215. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, Y.B.; Lao, M.Y.; Chen, W.; Hu, Q.D.; Zhi, X.; Chen, Z.L.; Bai, X.L.; Dang, X.W.; Liang, T.B. Endogenous Interleukin 18 Suppresses Liver Regeneration After Hepatectomy in Mice. Liver Transplant. 2020, 26, 408–418. [Google Scholar] [CrossRef]
- Liu, B.; Paranjpe, S.; Bowen, W.C.; Bell, A.W.; Luo, J.-H.; Yu, Y.-P.; Mars, W.M.; Michalopoulos, G.K. Investigation of the Role of Glypican 3 in Liver Regeneration and Hepatocyte Proliferation. Am. J. Pathol. 2009, 175, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Bell, A.W.; Paranjpe, S.; Bowen, W.C.; Khillan, J.S.; Luo, J.-H.; Mars, W.M.; Michalopoulos, G.K. Suppression of liver regeneration and hepatocyte proliferation in hepatocyte-targeted glypican 3 transgenic mice. Hepatology 2010, 52, 1060–1067. [Google Scholar] [CrossRef]
- Lin, C.-W.; Mars, W.M.; Paranjpe, S.; Donthamsetty, S.; Bhave, V.S.; Kang, L.-I.; Orr, A.; Bowen, W.C.; Bell, A.W.; Michalopoulos, G.K. Hepatocyte proliferation and hepatomegaly induced by phenobarbital and 1,4-bis [2-(3,5-dichloropyridyloxy)] benzene is suppressed in hepatocyte-targeted glypican 3 transgenic mice. Hepatology 2011, 54, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Margamuljana, L.; Joseph, C.; Schouteden, S.; Buckley, S.M.; Verfaillie, C.M. Glypican-3–mediated inhibition of CD26 by TFPI: A novel mechanism in hematopoietic stem cell homing and maintenance. Blood 2013, 121, 2587–2595. [Google Scholar] [CrossRef]
- Oikari, L.E.; Okolicsanyi, R.K.; Qin, A.; Yu, C.; Griffiths, L.R.; Haupt, L.M. Cell surface heparan sulfate proteoglycans as novel markers of human neural stem cell fate determination. Stem Cell Res. 2016, 16, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Emoto, M.; Okuya, S.; Fukuda, N.; Nakamori, Y.; Miyazaki, M.; Miyamoto, S.; Tanabe, K.; Aburatani, H.; Oka, Y.; et al. Identification of Glypican3 as a novel GLUT4-binding protein. Biochem. Biophys. Res. Commun. 2008, 369, 1204–1208. [Google Scholar] [CrossRef]
- Petit, L.M.G.; Cherif, L.S.; Devilliers, M.A.; Hatoum, S.; Ancel, J.; Delepine, G.; Durlach, A.; Dubernard, X.; Mérol, J.C.; Ruaux, C.; et al. Glypican-3 is a key tuner of the Hedgehog pathway in COPD. Heliyon 2025, 11, e41564. [Google Scholar] [CrossRef]
- Guo, S.; Chen, Q.; Liang, J.; Wu, H.; Li, L.; Wang, Y. Correlation of Glycolysis-immune-related Genes in the Follicular Microenvironment of Endometriosis Patients with ART Outcomes. Reprod. Sci. 2024, 31, 3357–3367. [Google Scholar] [CrossRef]
- Gokce, S.; Herkiloglu, D.; Cevik, O.; Turan, V. Evaluation of Intrafollicular Syndecan 1, Glypican 3, and Spermidine Levels in Women with Diminished Ovarian Reserve. Reprod. Sci. 2023, 30, 569–575. [Google Scholar] [CrossRef]
- Elliott, M.; Maher, E.R. Beckwith-Wiedemann syndrome. J. Med. Genet. 1994, 31, 560–564. [Google Scholar] [CrossRef]
- Weksberg, R.; Squire, J.A. Molecular biology of Beckwith-Wiedemann syndrome. Med. Pediatr. Oncol. 1996, 27, 462–469. [Google Scholar] [CrossRef]
- Xu, Y.; Papageorgiou, A.; Polychronakos, C. Developmental regulation of the soluble form of insulin-like growth factor-II mannose 6-phosphate receptor in human serum and amniotic fluid. J. Clin. Endocrinol. Metab. 1998, 83, 437–442. [Google Scholar]
- Weksberg, R.; Squire, J.A.; Templeton, D.M. Glypicans: A growing trend. Nat. Genet. 1996, 12, 225–227. [Google Scholar] [CrossRef]
- Song, H.H.; Shi, W.; Xiang, Y.Y.; Filmus, J. The loss of glypican-3 induces alterations in Wnt signaling. J. Biol. Chem. 2005, 280, 2116–2125. [Google Scholar] [CrossRef]
- Capurro, M.I.; Li, F.; Filmus, J. Overgrowth of a mouse model of Simpson–Golabi–Behmel syndrome is partly mediated by Indian Hedgehog. EMBO Rep. 2009, 10, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Xu, P.; Shi, W.; Li, F.; Jia, A.; Filmus, J. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev. Cell 2008, 14, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Bhave, V.S.; Mars, W.; Donthamsetty, S.; Zhang, X.; Tan, L.; Luo, J.; Bowen, W.C.; Michalopoulos, G.K. Regulation of Liver Growth by Glypican 3, CD81, Hedgehog, and Hhex. Am. J. Pathol. 2013, 183, 153–159. [Google Scholar] [CrossRef]
- Liu, Y.C.; Wierbowski, B.M.; Salic, A. Hedgehog pathway modulation by glypican 3-conjugated heparan sulfate. J. Cell Sci. 2022, 135, jcs259297. [Google Scholar] [CrossRef]
- Grisaru, S.; Cano-Gauci, D.; Tee, J.; Filmus, J.; Rosenblum, N.D. Glypican-3 Modulates BMP- and FGF-Mediated Effects during Renal Branching Morphogenesis. Dev. Biol. 2001, 231, 31–46. [Google Scholar] [CrossRef]
- Akkermans, O.; Delloye-Bourgeois, C.; Peregrina, C.; Carrasquero-Ordaz, M.; Kokolaki, M.; Berbeira-Santana, M.; Chavent, M.; Reynaud, F.; Raj, R.; Agirre, J.; et al. GPC3-Unc5 receptor complex structure and role in cell migration. Cell 2022, 185, 3931–3949.e26. [Google Scholar] [CrossRef]
- Vences-Catalán, F.; Duault, C.; Kuo, C.-C.; Rajapaksa, R.; Levy, R.; Levy, S. CD81 as a tumor target. Biochem. Soc. Trans. 2017, 45, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; Selfe, J.; Missiaglia, E.; Fisher, C.; Shipley, J. Glypican-3 is expressed in rhabdomyosarcomas but not adult spindle cell and pleomorphic sarcomas. J. Clin. Pathol. 2011, 64, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Shibui, Y.; Miyoshi, K.; Kohashi, K.; Kinoshita, Y.; Kuda, M.; Yamamoto, H.; Taguchi, T.; Oda, Y. Glypican-3 expression in malignant small round cell tumors. Oncol. Lett. 2019, 17, 3523–3528. [Google Scholar] [CrossRef]
- Zynger, D.L.; Dimov, N.D.; Luan, C.; Teh, B.T.; Yang, X.J. Glypican 3: A Novel Marker in Testicular Germ Cell Tumors. Am. J. Surg. Pathol. 2006, 30, 1570–1575. [Google Scholar] [CrossRef]
- Ho, M.; Kim, H. Glypican-3: A new target for cancer immunotherapy. Eur. J. Cancer 2011, 47, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, H.; Weng, H.; Zhang, X.; Li, P.; Fan, C.-L.; Li, B.; Dong, P.-L.; Li, L.; Dooley, S.; et al. Glypican-3 promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells through ERK signaling pathway. Int. J. Oncol. 2015, 46, 1275–1285. [Google Scholar] [CrossRef]
- Yamauchi, N.; Watanabe, A.; Hishinuma, M.; Ohashi, K.; Midorikawa, Y.; Morishita, Y.; Niki, T.; Shibahara, J.; Mori, M.; Makuuchi, M.; et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod. Pathol. 2005, 18, 1591–1598. [Google Scholar] [CrossRef]
- Aviel-Ronen, S.; Lau, S.K.; Pintilie, M.; Lau, D.; Liu, N.; Tsao, M.S.; Jothy, S. Glypican-3 is overexpressed in lung squamous cell carcinoma, but not in adenocarcinoma. Mod. Pathol. 2008, 21, 817–825. [Google Scholar] [CrossRef]
- Wang, D.; Gao, Y.; Zhang, Y.; Wang, L.; Chen, G. Glypican-3 promotes cell proliferation and tumorigenesis through up-regulation of beta-catenin expression in lung squamous cell carcinoma. Biosci. Rep. 2019, 39, BSR20181147. [Google Scholar] [CrossRef]
- Ning, J.; Jiang, S.; Li, X.; Wang, Y.; Deng, X.; Zhang, Z.; He, L.; Wang, D.; Jiang, Y. GPC3 affects the prognosis of lung adenocarcinoma and lung squamous cell carcinoma. BMC Pulm. Med. 2021, 21, 199. [Google Scholar]
- Pantanowitz, L.; Otis, C.N. Glypican-3 immunohistochemistry in the ovary. Histopathology 2008, 53, 115–117. [Google Scholar] [CrossRef]
- Zynger, D.L.; Everton, M.J.; Dimov, N.D.; Chou, P.M.; Yang, X.J. Expression of Glypican 3 in Ovarian and Extragonadal Germ Cell Tumors. Am. J. Clin. Pathol. 2008, 130, 224–230. [Google Scholar] [CrossRef]
- Sakurai, M.; Shibata, K.; Umezu, T.; Kajiyama, H.; Yamamoto, E.; Ino, K.; Nawa, A.; Kikkawa, F. Growth-suppressing function of glypican-3 (GPC3) via insulin like growth factor II (IGF-II) signaling pathway in ovarian clear cell carcinoma cells. Gynecol. Oncol. 2010, 119, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, S.; Gueth, U.; Baumhoer, D.; Moch, H.; Terracciano, L.; Singer, G. Glypican-3 Expression in Primary and Recurrent Ovarian Carcinomas. Int. J. Gynecol. Pathol. 2007, 26, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Nakatsura, T.; Kageshita, T.; Ito, S.; Wakamatsu, K.; Monji, M.; Ikuta, Y.; Senju, S.; Ono, T.; Nishimura, Y. Identification of glypican-3 as a novel tumor marker for melanoma. Clin. Cancer Res. 2004, 10, 6612–6621. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.S.; Shen, T.; De Rienzo, A.; Lee, W.C.; Ferriola, P.C.; Jhanwar, S.C.; Mossman, B.T.; Filmus, J.; Testa, J.R. Expression of GPC3, an X-linked recessive overgrowth gene, is silenced in malignant mesothelioma. Oncogene 2000, 19, 410–416. [Google Scholar] [CrossRef]
- Rahbari, M.; Pecqueux, M.; Aust, D.; Stephan, H.; Tiebel, O.; Chatzigeorgiou, A.; Tonn, T.; Baenke, F.; Rao, V.; Ziegler, N.; et al. Expression of Glypican 3 Is an Independent Prognostic Biomarker in Primary Gastro-Esophageal Adenocarcinoma and Corresponding Serum Exosomes. J. Clin. Med. 2019, 8, 696. [Google Scholar] [CrossRef]
- Ortiz, M.V.; Roberts, S.S.; Bender, J.G.; Shukla, N.; Wexler, L.H. Immunotherapeutic Targeting of GPC3 in Pediatric Solid Embryonal Tumors. Front. Oncol. 2019, 9, 108. [Google Scholar] [CrossRef]
- Yao, H.; Yang, Z.; Liu, Z.; Miao, X.; Yang, L.; Li, D.; Zou, Q.; Yuan, Y. Glypican-3 and KRT19 are markers associating with metastasis and poor prognosis of pancreatic ductal adenocarcinoma. Cancer Biomark. 2017, 17, 397–404. [Google Scholar] [CrossRef]
- Yamanaka, K.; Ito, Y.; Okuyama, N.; Noda, K.; Matsumoto, H.; Yoshida, H.; Miyauchi, A.; Capurro, M.; Filmus, J.; Miyoshi, E. Immunohistochemical study of glypican 3 in thyroid cancer. Oncology 2007, 73, 389–394. [Google Scholar] [CrossRef]
- Kim, H.; Xu, G.L.; Borczuk, A.C.; Busch, S.; Filmus, J.; Capurro, M.; Brody, J.S.; Lange, J.; D’Armiento, J.M.; Rothman, P.B.; et al. The heparan sulfate proteoglycan is a potential lung tumor suppressor. Am. J. Respir. Cell Mol. Biol. 2003, 29, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.Y.; Ladeda, V.; Filmus, J. Glypican-3 expression is silenced in human breast cancer. Oncogene 2001, 20, 7408–7412. [Google Scholar] [CrossRef]
- Adugna, A.; Amare, G.A.; Jemal, M. Current Advancements in Serum Protein Biomarkers for Hepatitis B Virus-Associated Hepatocyte Remodeling and Hepatocellular Carcinoma. Immun. Inflamm. Dis. 2025, 13, e70171. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, W.; Zhou, Y.; Tan, X.; Huang, Q.; Liang, J.; Zhou, Z. Label-free determination of glypican-3 using PtPd@H-rGO nanocomposites decorated light-addressable potentiometric sensor. Bioelectrochemistry 2025, 162, 108855. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, C.M.; Eilber, F.C.; Butterfield, L.H.; Ribas, A.; Dissette, V.B.; Koh, A.; Montejo, L.D.; Lee, M.C.; Andrews, K.J.; McBride, W.H.; et al. alpha-fetoprotein-specific genetic immunotherapy for hepatocellular carcinoma. Cancer Res. 1999, 59, 3064–3067. [Google Scholar]
- Cao, B.; Ni, Q.; Chen, Z.; Yang, S.; Zhang, X.; Su, H.; Zhang, Z.; Zhao, Q.; Zhu, X.; Liu, M. Development of glypican-3-specific chimeric antigen receptor-modified natural killer cells and optimization as a therapy for hepatocellular carcinoma. J. Leukoc. Biol. 2025, 117, qiae144. [Google Scholar] [CrossRef]
- Sui, M.; Liu, T.; Song, X.; Li, J.; Ding, H.; Liu, Y.; Wang, X.; Liu, H.; Xue, Y.; Qi, J.; et al. The molecular receptor NKBB enhances the persistence and anti-hepatocellular carcinoma activity of GPC3 CAR-T cells. Pharmacol. Res. 2025, 212, 107619. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.L.; Pan, Z.J.; Lei, C.J.; Wen, J.Y.; Li, M.Y.; Liu, Z.K.; Qiu, Z.D.; Lin, M.Z.; Chen, N.P.; Chen, M. Knockdown of GPC3 inhibits the proliferation of Huh7 hepatocellular carcinoma cells through down-regulation of YAP. J. Cell. Biochem. 2013, 114, 625–631. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Chen, W.; Zheng, P.; Liu, T.; He, W.; Zhang, J.; Zeng, X. Silencing glypican-3 expression induces apoptosis in human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 419, 656–661. [Google Scholar] [CrossRef]
- Sun, C.K.; Chua, M.-S.; He, J.; Samuel, K.S. Suppression of Glypican 3 Inhibits Growth of Hepatocellular Carcinoma Cells through Up-Regulation of TGF-beta 2. Neoplasia 2011, 13, 735–747. [Google Scholar] [CrossRef]
- Luo, R. Inhibition of glypican-3 expression via RNA interference influences the growth and invasive ability of the MHCC97-H human hepatocellular carcinoma cell line. Int. J. Mol. Med. 2011, 28, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Xiang, Y.Y.; Lobe, C.; Filmus, J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Qi, X.-H.; Wu, D.; Cui, H.-X.; Ma, N.; Su, J.; Wang, Y.-T.; Jiang, Y.-H. Silencing of the glypican-3 gene affects the biological behavior of human hepatocellular carcinoma cells. Mol. Med. Rep. 2014, 10, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Tseng, C.-J.; Lin, T.T.C.; Cheng, I.; Pan, H.-W.; Hsu, H.-C.; Lee, Y.-M. Glypican-3-mediated oncogenesis involves the Insulin-like growth factor-signaling pathway. Carcinogenesis 2008, 29, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Shibata, K.; Kajiyama, H.; Yamamoto, E.; Nawa, A.; Kikkawa, F. Glypican-3 expression predicts poor clinical outcome of patients with early-stage clear cell carcinoma of the ovary. J. Clin. Pathol. 2010, 63, 962–966. [Google Scholar] [CrossRef]
- Hu, R.; Zhu, Z. ELK1-activated GPC3-AS1/GPC3 axis promotes the proliferation and migration of cervical cancer cells. J. Gene Med. 2019, 21, e3099. [Google Scholar] [CrossRef]
- Ning, J.; Ding, J.; Wang, S.; Jiang, Y.; Wang, D.; Jiang, S. GPC3 Promotes Lung Squamous Cell Carcinoma Progression and HLA-A2-Restricted GPC3 Antigenic Peptide-Modified Dendritic Cell-Induced Cytotoxic T Lymphocytes to Kill Lung Squamous Cell Carcinoma Cells. J. Immunol. Res. 2023, 2023, 5532617. [Google Scholar] [CrossRef]
- Ma, H.-F.; Shu, P.; Shi, X.-H.; Wang, M.; Jiang, M.-F. Identification of miR-4510 as a metastasis suppressor of gastric cancer through regulation of tumor microenvironment via targeting GPC3. Clin. Exp. Metastasis 2022, 39, 363–374. [Google Scholar] [CrossRef]
- Wichert, A.; Stege, A.; Midorikawa, Y.; Holm, P.S.; Lage, H. Glypican-3 is involved in cellular protection against mitoxantrone in gastric carcinoma cells. Oncogene 2003, 23, 945–955. [Google Scholar] [CrossRef]
- Kandil, D.H.; Cooper, K. Glypican-3 A Novel Diagnostic Marker for Hepatocellular Carcinoma and More. Adv. Anat. Pathol. 2009, 16, 125–129. [Google Scholar] [CrossRef]
- Akutsu, N.; Yamamoto, H.; Sasaki, S.; Taniguchi, H.; Arimura, Y.; Imai, K.; Shinomura, Y. Association of glypican-3 expression with growth signaling molecules in hepatocellular carcinoma. World J. Gastroentero 2010, 16, 3521–3528. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Tang, Z.W.; Zhang, Y.F.; Feng, M.Q.; Qian, M.; Dimitrov, D.S.; Ho, M. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat. Commun. 2015, 6, 6536. [Google Scholar] [CrossRef]
- Cheng, W.; Huang, P.C.; Chao, H.M.; Jeng, Y.M.; Hsu, H.C.; Pan, H.W.; Hwu, W.L.; Lee, Y.M. Glypican-3 induces oncogenicity by preventing IGF-1R degradation, a process that can be blocked by Grb10. Oncotarget 2017, 8, 80429–80442. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Gao, W.; Wang, R.; Chen, W.; Man, Y.-G.; Figg, W.D.; Wang, X.W.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, E1083–E1091. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Shi, C.; Fu, S.; Sun, Y.-F.; Li, C. Targeting GPC3 high cancer-associated fibroblasts sensitizing the PD-1 blockage therapy in gastric cancer. Ann. Med. 2023, 55, 2189295. [Google Scholar] [CrossRef]
- Lai, J.-P.; Sandhu, D.S.; Yu, C.; Han, T.; Moser, C.D.; Jackson, K.K.; Guerrero, R.B.; Aderca, I.; Isomoto, H.; Garrity-Park, M.M.; et al. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology 2008, 47, 1211–1222. [Google Scholar] [CrossRef]
- Wong, C.-M.; Gao, W.; Kim, H.; Ho, M. Human Monoclonal Antibody Targeting the Heparan Sulfate Chains of Glypican-3 Inhibits HGF-Mediated Migration and Motility of Hepatocellular Carcinoma Cells. PLoS ONE 2015, 10, e0137664. [Google Scholar]
- Lin, H.C.; Huber, R.; Schlessinger, D.; Morin, P.J. Frequent silencing of the GPC3 gene in ovarian cancer cell lines. Cancer Res. 1999, 59, 807–810. [Google Scholar]
- Imon, R.R.; Aktar, S.; Morshed, N.; Nur, S.M.; Mahtarin, R.; Rahman, F.A.; Talukder, M.E.K.; Alam, R.; Karpiński, T.M.; Ahammad, F.; et al. Biological and clinical significance of the glypican-3 gene in human lung adenocarcinoma: An in silico analysis. Medicine 2023, 102, e35347. [Google Scholar] [CrossRef]
- Valsechi, M.C.; Oliveira, A.B.B.; Conceiçao, A.L.G.; Stuqui, B.; Candido, N.M.; Provazzi, P.J.S.; de Araújo, L.F.; Silva, W.; Calmon, M.D.; Rahal, P. GPC3 reduces cell proliferation in renal carcinoma cell lines. BMC Cancer 2014, 14, 631. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.G.; Farías, E.; Colombo, L.; Filmus, J.; Puricelli, L.; Joffé, E.B.D. Inhibition of invasion and metastasis by glypican-3 in a syngeneic breast cancer model. Breast Cancer Res. Treat. 2003, 80, 221–232. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, D.; Liu, M.; Bai, J.; Zhou, X.; Gong, B.; Lu, J.; Zhang, Y.; Huang, H.; Luo, W.; et al. Downregulation of glypican-3 expression increases migration, invasion, and tumorigenicity of human ovarian cancer cells. Tumor Biol. 2015, 36, 7997–8006. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Zhang, J.; Zhao, Y.; Sun, M.; Zhao, H.; Li, S. miR-96 promotes invasion and metastasis by targeting GPC3 in non-small cell lung cancer cells. Oncol. Lett. 2018, 15, 9081–9086. [Google Scholar] [CrossRef]
- Varma, R.R.; Hector, S.M.; Clark, K.; Greco, W.R.; Hawthorn, L.; Pendyala, L. Gene expression profiling of a clonal isolate of oxaliplatin resistant ovarian carcinoma cell line A2780/C10. Oncol. Rep. 2005, 14, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Kwack, M.H.; Choi, B.Y.; Sung, Y.K. Cellular changes resulting from forced expression of glypican-3 in hepatocellular carcinoma cells. Mol. Cells 2006, 21, 224–228. [Google Scholar] [CrossRef]
- Farooq, M.; Hwang, S.Y.; Park, M.K.; Kim, J.C.; Kim, M.K.; Sung, Y.K. Blocking endogenous glypican-3 expression releases Hep 3B cells from G1 arrest. Mol. Cells 2003, 15, 356–360. [Google Scholar] [CrossRef]
- Han, S.W.; Ma, X.M.; Zhao, Y.X.; Zhao, H.Y.; Batista, A.; Zhou, S.; Zhou, X.N.; Yang, Y.; Wang, T.T.; Bi, J.T.; et al. Identification of Glypican-3 as a potential metastasis suppressor gene in gastric cancer. Oncotarget 2016, 7, 44406–44416. [Google Scholar] [CrossRef]
- Fernandez, D.; Guereno, M.; Huvelle, M.A.L.; Cercato, M.; Peters, M.G. Signaling network involved in the GPC3-induced inhibition of breast cancer progression: Role of canonical Wnt pathway. J. Cancer Res. Clin. Oncol. 2018, 144, 2399–2418. [Google Scholar] [CrossRef]
- Guereno, M.; Pastore, M.D.; Lugones, A.C.; Cercato, M.; Todaro, L.; Urtreger, A.; Peters, M.G. Glypican-3 (GPC3) inhibits metastasis development promoting dormancy in breast cancer cells by p38 MAPK pathway activation. Eur. J. Cell Biol. 2020, 99, 151096. [Google Scholar] [CrossRef]
- Capurro, M.I.; Shi, W.; Filmus, J. LRP1 mediates Hedgehog-induced endocytosis of the GPC3-Hedgehog complex. J. Cell Sci. 2012, 125, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.; Shi, W.; Izumikawa, T.; Kitagawa, H.; Filmus, J. Processing by convertases is required for glypican-3-induced inhibition of Hedgehog signaling. J. Biol. Chem. 2015, 290, 7576–7585. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wei, L.; Liu, X.; Bai, H.; Ye, Y.; Li, D.; Li, N.; Baxa, U.; Wang, Q.; Lv, L.; et al. A Frizzled-Like Cysteine-Rich Domain in Glypican-3 Mediates Wnt Binding and Regulates Hepatocellular Carcinoma Tumor Growth in Mice. Hepatology 2019, 70, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.; Martin, T.; Shi, W.; Filmus, J. Glypican-3 binds to Frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J. Cell Sci. 2014, 127, 1565–1575. [Google Scholar] [CrossRef]
- Gao, W.; Kim, H.; Feng, M.; Phung, Y.; Xavier, C.P.; Rubin, J.S.; Ho, M. Inactivation of Wnt Signaling by a Human Antibody That Recognizes the Heparan Sulfate Chains of Glypican-3 for Liver Cancer Therapy. Hepatology 2014, 60, 576–587. [Google Scholar] [CrossRef]
- Song, H.H.; Shi, W.; Filmus, J. OCI-5/Rat Glypican-3 Binds to Fibroblast Growth Factor-2 but Not to Insulin-like Growth Factor-2. J. Biol. Chem. 1997, 272, 7574–7577. [Google Scholar] [CrossRef]
- Zittermann, S.I.; Capurro, M.I.; Shi, W.; Filmus, J. Soluble glypican 3 inhibits the growth of hepatocellular carcinoma in vitro and in vivo. Int. J. Cancer 2010, 126, 1291–1301. [Google Scholar] [CrossRef]
- Davoodi, J.; Kelly, J.; Gendron, N.H.; MacKenzie, A.E. The Simpson–Golabi–Behmel syndrome causative Glypican-3, binds to and inhibits the dipeptidyl peptidase activity of CD26. Proteomics 2007, 7, 2300–2310. [Google Scholar] [CrossRef]

| Type of Cancer | Tumor vs. Normal | Reference |
|---|---|---|
| Rhabdomyosarcoma | high | [52,53] |
| Choriocarcinoma | high | [54] |
| Hepatocellular carcinoma | high | [55,56,57] |
| Lung squamous carcinoma | high | [58,59,60] |
| Yolk sac tumors | high | [61,62] |
| Ovarian clear cell carcinoma | high | [63,64] |
| Melanoma | high | [65] |
| Malignant mesothelioma | high | [66] |
| Gastro-esophageal adenocarcinoma | high | [67] |
| Pediatric solid embryonal tumors | high | [68] |
| Pancreatic ductal adenocarcinoma | high | [69] |
| Thyroid cancer | high | [70] |
| Lung adenocarcinoma | low | [71] |
| Breast cancer | low | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piao, Q.; Bian, X.; Zhao, Q.; Sun, L. Unraveling Glypican-3: From Structural to Pathophysiological Roles and Mechanisms—An Integrative Perspective. Cells 2025, 14, 726. https://doi.org/10.3390/cells14100726
Piao Q, Bian X, Zhao Q, Sun L. Unraveling Glypican-3: From Structural to Pathophysiological Roles and Mechanisms—An Integrative Perspective. Cells. 2025; 14(10):726. https://doi.org/10.3390/cells14100726
Chicago/Turabian StylePiao, Qianling, Xiaona Bian, Qi Zhao, and Luguo Sun. 2025. "Unraveling Glypican-3: From Structural to Pathophysiological Roles and Mechanisms—An Integrative Perspective" Cells 14, no. 10: 726. https://doi.org/10.3390/cells14100726
APA StylePiao, Q., Bian, X., Zhao, Q., & Sun, L. (2025). Unraveling Glypican-3: From Structural to Pathophysiological Roles and Mechanisms—An Integrative Perspective. Cells, 14(10), 726. https://doi.org/10.3390/cells14100726