


The Metabolic Landscape of Cancer Stem Cells: Insights and Implications for Therapy

 , , ,
, , ,  , ,
, ,  and
and
Abstract
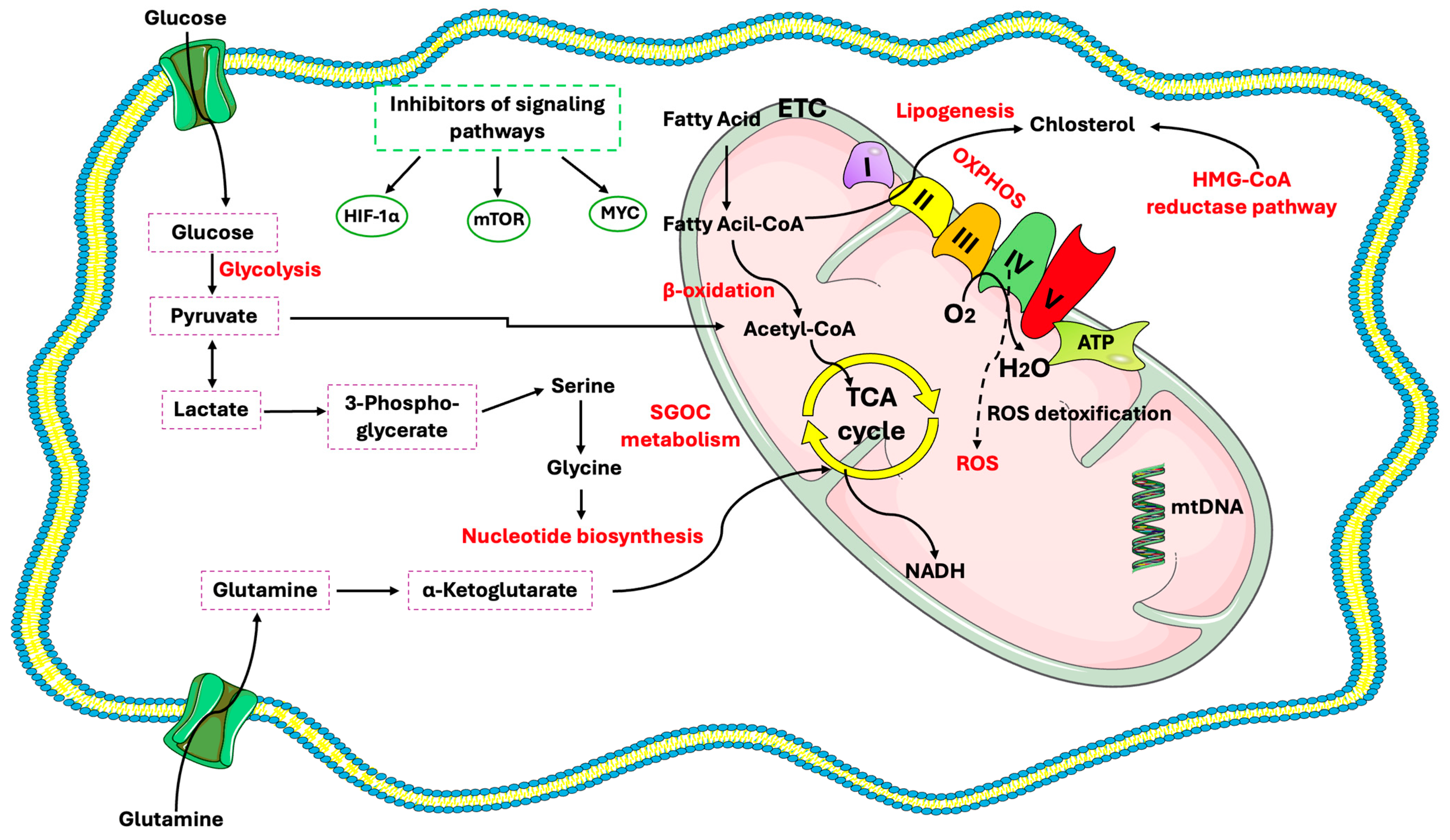
1. Introduction
2. CSC Characteristics
3. Glucose Metabolism in CSCs
Mitochondria in CSCs
4. Lipid Metabolism in CSCs
4.1. Key Lipid Regulators
4.2. Lipid Droplets
5. Regulation of Amino Acids Metabolism in CSCs
6. Metabolic Plasticity of CSC Within the TME
7. Epigenetic Regulation of Metabolic Pathways in CSCs
8. Optimizing Strategies for CSC Targeting
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Autorino, R.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Cancer Stem Cells in Renal Cell Carcinoma: Origins and Biomarkers. Int. J. Mol. Sci. 2023, 24, 13179. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S. Cancer Stem Cell Markers and Related Network Pathways; Springer International Publishing: Cham, Switzerland, 2022; Volume 1393, ISBN 978-3-031-12973-5. [Google Scholar]
- Kolios, G.; Moodley, Y. Introduction to Stem Cells and Regenerative Medicine. Respiration 2013, 85, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Tian, W.; Ning, J.; Xiao, G.; Zhou, Y.; Wang, Z.; Zhai, Z.; Tanzhu, G.; Yang, J.; Zhou, R. Cancer Stem Cells: Advances in Knowledge and Implications for Cancer Therapy. Signal Transduct. Target. Ther. 2024, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Bisht, S.; Nigam, M.; Kunjwal, S.S.; Sergey, P.; Mishra, A.P.; Sharifi-Rad, J. Cancer Stem Cells: From an Insight into the Basics to Recent Advances and Therapeutic Targeting. Stem Cells Int. 2022, 2022, 9653244. [Google Scholar] [CrossRef]
- Denham, M.; Conley, B.; Olsson, F.; Cole, T.J.; Mollard, R. Stem Cells: An Overview. Curr. Protoc. Cell Biol. 2005, 28. [Google Scholar] [CrossRef]
- Vats, A.; Bielby, R.; Tolley, N.; Nerem, R.; Polak, J. Stem Cells. Lancet 2005, 366, 592–602. [Google Scholar] [CrossRef]
- Shenghui, H.; Nakada, D.; Morrison, S.J. Mechanisms of Stem Cell Self-Renewal. Annu. Rev. Cell Dev. Biol. 2009, 25, 377–406. [Google Scholar] [CrossRef]
- Falanga, V. Stem Cells in Tissue Repair and Regeneration. J. Investig. Dermatol. 2012, 132, 1538–1541. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Stem Cell Metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the Cancer Stem Cells—What Challenges Do They Pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Gallardo, C.; Simón, C. Cells, Stem Cells, and Cancer Stem Cells. Semin. Reprod. Med. 2013, 31, 005–013. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of Cancer Stem Cell Metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer Stem Cell Metabolism: A Potential Target for Cancer Therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef]
- Yasuda, T.; Ishimoto, T.; Baba, H. Conflicting Metabolic Alterations in Cancer Stem Cells and Regulation by the Stromal Niche. Regen. Ther. 2021, 17, 8–12. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating Cancer Stem Cells—An Integrated Concept of Malignant Tumour Progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Moriyama, T.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Ikenaga, N.; Sato, N.; Tanaka, M. Enhanced Cell Migration and Invasion of CD133+ Pancreatic Cancer Cells Cocultured with Pancreatic Stromal Cells. Cancer 2010, 116, 3357–3368. [Google Scholar] [CrossRef]
- Fillmore, C.M.; Gupta, P.B.; Rudnick, J.A.; Caballero, S.; Keller, P.J.; Lander, E.S.; Kuperwasser, C. Estrogen Expands Breast Cancer Stem-like Cells through Paracrine FGF/Tbx3 Signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21737–21742. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Clonal and Population Analyses Demonstrate That an EGF-Responsive Mammalian Embryonic CNS Precursor Is a Stem Cell. Dev. Biol. 1996, 175, 1–13. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Vavallo, A.; Ditonno, P.; Battaglia, M. Isolation and Characterization of Cancer Stem Cells in Renal Cell Carcinoma. Urol. J. 2015, 82, 46–53. [Google Scholar] [CrossRef]
- Galleggiante, V.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Ditonno, P.; Bettocchi, C.; Selvaggi, F.P.; Lucarelli, G.; Battaglia, M. CTR2 Identifies a Population of Cancer Cells with Stem Cell-like Features in Patients with Clear Cell Renal Cell Carcinoma. J. Urol. 2014, 192, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Yamashita, S.; Ushijima, T.; Arai, T. Systemic Administration of Small Interfering RNA Targeting Human Nestin Inhibits Pancreatic Cancer Cell Proliferation and Metastasis. Pancreas 2016, 45, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Bhuvanalakshmi, G.; Arfuso, F.; Rajan, G.; Millward, M.; Dharmarajan, A. Cancer Stem-like Cells from Head and Neck Cancers Are Chemosensitized by the Wnt Antagonist, sFRP4, by Inducing Apoptosis, Decreasing Stemness, Drug Resistance and Epithelial to Mesenchymal Transition. Cancer Gene Ther. 2014, 21, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Karsten, U.; Goletz, S. What Makes Cancer Stem Cell Markers Different? SpringerPlus 2013, 2, 301. [Google Scholar] [CrossRef]
- Tomao, F.; Papa, A.; Rossi, L.; Strudel, M.; Vici, P.; Lo Russo, G.; Tomao, S. Emerging Role of Cancer Stem Cells in the Biology and Treatment of Ovarian Cancer: Basic Knowledge and Therapeutic Possibilities for an Innovative Approach. J. Exp. Clin. Cancer Res. 2013, 32, 48. [Google Scholar] [CrossRef]
- Moitra, K.; Lou, H.; Dean, M. Multidrug Efflux Pumps and Cancer Stem Cells: Insights Into Multidrug Resistance and Therapeutic Development. Clin. Pharmacol. Ther. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Ju, F.; Atyah, M.M.; Horstmann, N.; Gul, S.; Vago, R.; Bruns, C.J.; Zhao, Y.; Dong, Q.-Z.; Ren, N. Characteristics of the Cancer Stem Cell Niche and Therapeutic Strategies. Stem Cell Res. Ther. 2022, 13, 233. [Google Scholar] [CrossRef]
- Pan, Y.; Yuan, C.; Zeng, C.; Sun, C.; Xia, L.; Wang, G.; Chen, X.; Zhang, B.; Liu, J.; Ding, Z. Cancer Stem Cells and Niches: Challenges in Immunotherapy Resistance. Mol. Cancer 2025, 24, 52. [Google Scholar] [CrossRef]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic Insights into Pathophysiological Mechanisms and Biomarker Discovery in Clear Cell Renal Cell Carcinoma. Expert Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic Profile of Glycolysis and the Pentose Phosphate Pathway Identifies the Central Role of Glucose-6-Phosphate Dehydrogenase in Clear Cell-Renal Cell Carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer Stem Cells (CSCs): Metabolic Strategies for Their Identification and Eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy Metabolism of Cancer: Glycolysis versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Pascual, G.; Domínguez, D.; Benitah, S.A. The Contributions of Cancer Cell Metabolism to Metastasis. Dis. Models Mech. 2018, 11, dmm032920. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Lepore Signorile, M.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated Multi-Omics Characterization Reveals a Distinctive Metabolic Signature and the Role of NDUFA4L2 in Promoting Angiogenesis, Chemoresistance, and Mitochondrial Dysfunction in Clear Cell Renal Cell Carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Garber, A.J.; Karl, I.E.; Kipnis, D.M. Alanine and Glutamine Synthesis and Release from Skeletal Muscle. II. The Precursor Role of Amino Acids in Alanine and Glutamine Synthesis. J. Biol. Chem. 1976, 251, 836–843. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Sun, H.; Xu, L. The Pathogenesis and Therapeutic Implications of Metabolic Reprogramming in Renal Cell Carcinoma. Cell Death Discov. 2025, 11, 186. [Google Scholar] [CrossRef]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and Cellular Regulation of Glucose Transporter (GLUT) Proteins in Cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient Transporters in Cancer: Relevance to Warburg Hypothesis and Beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Okuda, H.; Kobayashi, A.; Watabe, K. Metabolic Genes in Cancer: Their Roles in Tumor Progression and Clinical Implications. Biochim. Biophys. Acta BBA-Rev. Cancer 2010, 1805, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Bhattacharyya, S. Cancer Stem Cells: Metabolic Characterization for Targeted Cancer Therapy. Front. Oncol. 2021, 11, 756888. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the Facilitative Glucose Transporter GLUT1 Inhibits the Self-Renewal and Tumor-Initiating Capacity of Cancer Stem Cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast Cancer Stem Cells Rely on Fermentative Glycolysis and Are Sensitive to 2-Deoxyglucose Treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian Cancer Spheroid Cells with Stem Cell-Like Properties Contribute to Tumor Generation, Metastasis and Chemotherapy Resistance through Hypoxia-Resistant Metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef]
- Liu, P.-P.; Liao, J.; Tang, Z.-J.; Wu, W.-J.; Yang, J.; Zeng, Z.-L.; Hu, Y.; Wang, P.; Ju, H.-Q.; Xu, R.-H.; et al. Metabolic Regulation of Cancer Cell Side Population by Glucose through Activation of the Akt Pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy Metabolism Characterization of a Novel Cancer Stem Cell- L Ike Line 3 AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, E.; Tsukahara, T.; Emori, M.; Murata, K.; Akamatsu, A.; Shibayama, Y.; Hamada, S.; Watanabe, Y.; Kaya, M.; Hirohashi, Y.; et al. Osteosarcoma-initiating Cells Show High Aerobic Glycolysis and Attenuation of Oxidative Phosphorylation Mediated by LIN28B. Cancer Sci. 2020, 111, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Gentles, A.; Nair, R.V.; Huang, M.; Lin, Y.; Lee, C.Y.; Cai, S.; Scheeren, F.A.; Kuo, A.H.; Diehn, M. Targeting Unique Metabolic Properties of Breast Tumor Initiating Cells. Stem Cells 2014, 32, 1734–1745. [Google Scholar] [CrossRef]
- Song, K.; Kwon, H.; Han, C.; Zhang, J.; Dash, S.; Lim, K.; Wu, T. Active Glycolytic Metabolism in CD133(+) Hepatocellular Cancer Stem Cells: Regulation by MIR-122. Oncotarget 2015, 6, 40822–40835. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and Lactate Increase Cancer Cell “Stemness,” Driving Recurrence, Metastasis and Poor Clinical Outcome in Breast Cancer: Achieving Personalized Medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-Activated Macrophages Induced Aerobic Glycolysis and Epithelial-Mesenchymal Transition in Breast Cancer by Regulation of CCL5-CCR5 Axis: A Positive Metabolic Feedback Loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic Heterogeneity Confers Differences in Melanoma Metastatic Potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Lasorsa, F.; Di Meo, N.A.; Rutigliano, M.; Ferro, M.; Terracciano, D.; Tataru, O.S.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Emerging Hallmarks of Metabolic Reprogramming in Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 910. [Google Scholar] [CrossRef]
- Yuen, C.A.; Asuthkar, S.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. Cancer Stem Cell Molecular Reprogramming of the Warburg Effect in Glioblastomas: A New Target Gleaned from an Old Concept. CNS Oncol. 2016, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-W.; Tong, G.-H.; Liu, Y. Cancer Stem Cells and Hypoxia-Inducible Factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef]
- Wang, S.-F.; Tseng, L.-M.; Lee, H.-C. Role of Mitochondrial Alterations in Human Cancer Progression and Cancer Immunity. J. Biomed. Sci. 2023, 30, 61. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; López-Calcerrada, S.; Sierra-Magro, A.; Pérez-Pérez, R.; Formosa, L.E.; Hock, D.H.; Illescas, M.; Peñas, A.; Brischigliaro, M.; Ding, S.; et al. Two Independent Respiratory Chains Adapt OXPHOS Performance to Glycolytic Switch. Cell Metab. 2022, 34, 1792–1808.e6. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial Biogenesis Is Required for the Anchorage-Independent Survival and Propagation of Stem-like Cancer Cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef]
- Wen, G.-M.; Xu, X.-Y.; Xia, P. Metabolism in Cancer Stem Cells: Targets for Clinical Treatment. Cells 2022, 11, 3790. [Google Scholar] [CrossRef]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer Stem Cells from Epithelial Ovarian Cancer Patients Privilege Oxidative Phosphorylation, and Resist Glucose Deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid Cancer Stem Cells Display Reprogrammed Metabolism and Obtain Energy by Actively Running the Tricarboxylic Acid (TCA) Cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 Controls Oxidative Phosphorylation and Is Crucial for Preserving Glioblastoma Cancer Stem Cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene Ablation-Resistant Pancreatic Cancer Cells Depend on Mitochondrial Function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of Peroxisome-proliferator Activated Receptor-gamma (PPARγ) and the PPARγ Co-activator, PGC-1, in Human Breast Cancer Correlates with Clinical Outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as New Therapeutic Targets for Eradicating Cancer Stem Cells: Quantitative Proteomics and Functional Validation via MCT1/2 Inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyari, Y.; Dratver, M.B.; Pajonk, F. Metabolic Differences in Breast Cancer Stem Cells and Differentiated Progeny. Breast Cancer Res. Treat. 2014, 146, 525–534. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High Mitochondrial Mass Identifies a Sub-Population of Stem-like Cancer Cells That Are Chemo-Resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Battaglia, M.; Ditonno, P.; Lucarelli, G. The Dark Side of Lipid Metabolism in Prostate and Renal Carcinoma: Novel Insights into Molecular Diagnostic and Biomarker Discovery. Expert Rev. Mol. Diagn. 2023, 23, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of Lipidomics and Transcriptomics Reveals Reprogramming of the Lipid Metabolism and Composition in Clear Cell Renal Cell Carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Ferro, M.; Terracciano, D.; Buonerba, C.; Lucarelli, G.; Bottero, D.; Perdonà, S.; Autorino, R.; Serino, A.; Cantiello, F.; Damiano, R.; et al. The Emerging Role of Obesity, Diet and Lipid Metabolism in Prostate Cancer. Future Oncol. 2017, 13, 285–293. [Google Scholar] [CrossRef]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The Glucose and Lipid Metabolism Reprogramming Is Grade-Dependent in Clear Cell Renal Cell Carcinoma Primary Cultures and Is Targetable to Modulate Cell Viability and Proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Delmas, D.; Mialhe, A.; Cotte, A.K.; Connat, J.-L.; Bouyer, F.; Hermetet, F.; Aires, V. Lipid Metabolism in Cancer: Exploring Phospholipids as Potential Biomarkers. Biomed. Pharmacother. 2025, 187, 118095. [Google Scholar] [CrossRef]
- Mordzińska-Rak, A.; Verdeil, G.; Hamon, Y.; Błaszczak, E.; Trombik, T. Dysregulation of Cholesterol Homeostasis in Cancer Pathogenesis. Cell. Mol. Life Sci. 2025, 82, 168. [Google Scholar] [CrossRef]
- Fu, W.; Sun, A.; Dai, H. Lipid Metabolism Involved in Progression and Drug Resistance of Breast Cancer. Genes Dis. 2025, 12, 101376. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid Rafts as Major Platforms for Signaling Regulation in Cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; Pandolfi, P. A PML-PPARδ Pathway for Fatty Acid Oxidation Regulates Haematopoietic Stem Cell Maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- de Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E. Intratumor Cholesteryl Ester Accumulation Is Associated with Human Breast Cancer Proliferation and Aggressive Potential: A Molecular and Clinicopathological Study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef]
- Mitra, R.; Chao, O.; Urasaki, Y.; Goodman, O.B.; Le, T.T. Detection of Lipid-Rich Prostate Circulating Tumour Cells with Coherent Anti-Stokes Raman Scattering Microscopy. BMC Cancer 2012, 12, 540. [Google Scholar] [CrossRef]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid Droplets: A New Player in Colorectal Cancer Stem Cells Unveiled by Spectroscopic Imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef]
- Peck, B.; Schug, Z.T.; Zhang, Q.; Dankworth, B.; Jones, D.T.; Smethurst, E.; Patel, R.; Mason, S.; Jiang, M.; Saunders, R.; et al. Inhibition of Fatty Acid Desaturation Is Detrimental to Cancer Cell Survival in Metabolically Compromised Environments. Cancer Metab. 2016, 4, 6. [Google Scholar] [CrossRef]
- La Civita, E.; Sirica, R.; Crocetto, F.; Ferro, M.; Lasorsa, F.; Lucarelli, G.; Imbimbo, C.; Formisano, P.; Beguinot, F.; Terracciano, D. FABP4-Mediated ERK Phosphorylation Promotes Renal Cancer Cell Migration. BMC Cancer 2025, 25, 575. [Google Scholar] [CrossRef]
- Kawahara, I.; Mori, T.; Goto, K.; Fujii, K.; Ohmori, H.; Kishi, S.; Fujiwara-Tani, R.; Kuniyasu, H. Fatty Acids Induce Stemness in the Stromal Cells of a CT26 Mouse Tumor Model. Pathobiology 2017, 84, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-C.; Ntambi, J.M. Regulation of Stearoyl-CoA Desaturase Genes: Role in Cellular Metabolism and Preadipocyte Differentiation. Biochem. Biophys. Res. Commun. 1999, 266, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; Perin, L.; Bottazzi, B.; Mantovani, A.; Giavazzi, R.; Salmona, M. Membrane Fluidity Affects Tumor-cell Motility, Invasion and Lung-colonizing Potential. Int. J. Cancer 1989, 44, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-Desaturase 1 Regulates Lung Cancer Stemness via Stabilization and Nuclear Localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Castro, L.F.C.; Wilson, J.M.; Gonçalves, O.; Galante-Oliveira, S.; Rocha, E.; Cunha, I. The Evolutionary History of the Stearoyl-CoA Desaturase Gene Family in Vertebrates. BMC Evol. Biol. 2011, 11, 132. [Google Scholar] [CrossRef]
- Bansal, S.; Berk, M.; Alkhouri, N.; Partrick, D.A.; Fung, J.J.; Feldstein, A. Stearoyl-CoA Desaturase Plays an Important Role in Proliferation and Chemoresistance in Human Hepatocellular Carcinoma. J. Surg. Res. 2014, 186, 29–38. [Google Scholar] [CrossRef]
- Lai, K.K.Y.; Kweon, S.-M.; Chi, F.; Hwang, E.; Kabe, Y.; Higashiyama, R.; Qin, L.; Yan, R.; Wu, R.P.; Lai, K.; et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology 2017, 152, 1477–1491. [Google Scholar] [CrossRef]
- Colacino, J.A.; McDermott, S.P.; Sartor, M.A.; Wicha, M.S.; Rozek, L.S. Transcriptomic Profiling of Curcumin-Treated Human Breast Stem Cells Identifies a Role for Stearoyl-Coa Desaturase in Breast Cancer Prevention. Breast Cancer Res. Treat. 2016, 158, 29–41. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.-P.; Wang, B.; Cohen, A.; et al. SCD1 Inhibition Causes Cancer Cell Death by Depleting Mono-Unsaturated Fatty Acids. PLoS ONE 2012, 7, e33823. [Google Scholar] [CrossRef]
- Pandey, P.R.; Xing, F.; Sharma, S.; Watabe, M.; Pai, S.K.; Iiizumi-Gairani, M.; Fukuda, K.; Hirota, S.; Mo, Y.-Y.; Watabe, K. Elevated Lipogenesis in Epithelial Stem-like Cell Confers Survival Advantage in Ductal Carcinoma in Situ of Breast Cancer. Oncogene 2013, 32, 5111–5122. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-Regulated Lipid Metabolism: Convergent Physiology—Divergent Pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, W.; Luo, M.; Zhou, Y.; Chang, G.; Ren, W.; Wu, K.; Li, X.; Shen, J.; Zhao, X.; et al. SREBP1 Regulates Tumorigenesis and Prognosis of Pancreatic Cancer through Targeting Lipid Metabolism. Tumor Biol. 2015, 36, 4133–4141. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Z.; Song, L.; Gao, J.; Liu, Y. Lipid Metabolism of Cancer Stem Cells (Review). Oncol. Lett. 2022, 23, 119. [Google Scholar] [CrossRef]
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate Metabolism Regulates Basal Breast Cancer Stem Cells and Is a Potential Therapeutic Target. Stem Cells 2012, 30, 1327–1337. [Google Scholar] [CrossRef]
- Ganguly, K.; Rauth, S.; Marimuthu, S.; Kumar, S.; Batra, S.K. Unraveling Mucin Domains in Cancer and Metastasis: When Protectors Become Predators. Cancer Metastasis Rev. 2020, 39, 647–659. [Google Scholar] [CrossRef]
- Liu, C.; Chen, H.; Hu, B.; Shi, J.; Chen, Y.; Huang, K. New Insights into the Therapeutic Potentials of Statins in Cancer. Front. Pharmacol. 2023, 14, 1188926. [Google Scholar] [CrossRef]
- Ediriweera, M.K. Use of Cholesterol Metabolism for Anti-Cancer Strategies. Drug Discov. Today 2022, 27, 103347. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The Interplay between Cell Signalling and the Mevalonate Pathway in Cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Lucarelli, G.; Loizzo, D.; Ferro, M.; Rutigliano, M.; Vartolomei, M.D.; Cantiello, F.; Buonerba, C.; Di Lorenzo, G.; Terracciano, D.; De Cobelli, O.; et al. Metabolomic Profiling for the Identification of Novel Diagnostic Markers and Therapeutic Targets in Prostate Cancer: An Update. Expert Rev. Mol. Diagn. 2019, 19, 377–387. [Google Scholar] [CrossRef]
- Di Lorenzo, G.; Sonpavde, G.; Pond, G.; Lucarelli, G.; Rossetti, S.; Facchini, G.; Scagliarini, S.; Cartenì, G.; Federico, P.; Daniele, B.; et al. Statin Use and Survival in Patients with Metastatic Castration-Resistant Prostate Cancer Treated with Abiraterone Acetate. Eur. Urol. Focus 2018, 4, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A.; Buonerba, C.; Pond, G.; Crona, D.; Gillessen, S.; Lucarelli, G.; Rossetti, S.; Dorff, T.; Artale, S.; Locke, J.A.; et al. Statin Use and Survival in Patients with Metastatic Castration-Resistant Prostate Cancer Treated with Abiraterone or Enzalutamide after Docetaxel Failure: The International Retrospective Observational STABEN Study. Oncotarget 2018, 9, 19861–19873. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Pagliari, F.; Limongi, T.; Marini, M.; Falqui, A.; Seco, J.; Candeloro, P.; Liberale, C.; Di Fabrizio, E. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells Int. 2017, 2017, 1656053. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Li, J.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Peng, S.; Li, X.; et al. Emerging Role of Lipid Metabolism Alterations in Cancer Stem Cells. J. Exp. Clin. Cancer Res. 2018, 37, 118. [Google Scholar] [CrossRef]
- Ma, Z.; Pan, S.; Yang, Y.; Ren, H.; Yin, S.; Chen, Q.; An, Z.; Zhao, X.; Xu, Z. Lipid Droplets: Emerging Therapeutic Targets for Age-Related Metabolic Diseases. Ageing Res. Rev. 2025, 108, 102758. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-kDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef]
- Maan, M.; Peters, J.M.; Dutta, M.; Patterson, A.D. Lipid Metabolism and Lipophagy in Cancer. Biochem. Biophys. Res. Commun. 2018, 504, 582–589. [Google Scholar] [CrossRef]
- Lue, H.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.-H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic Reprogramming Ensures Cancer Cell Survival despite Oncogenic Signaling Blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Kuzmin, A.N.; Pliss, A.; Yadav, N.; Balachandar, S.; Wang, J.; Attwood, K.; Prasad, P.N.; Chandra, D. Lipid Quantification by Raman Microspectroscopy as a Potential Biomarker in Prostate Cancer. Cancer Lett. 2017, 397, 52–60. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF Drives Lipid Deposition and Cancer in ccRCC via Repression of Fatty Acid Metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed]
- Menard, J.A.; Christianson, H.C.; Kucharzewska, P.; Bourseau-Guilmain, E.; Svensson, K.J.; Lindqvist, E.; Chandran, V.I.; Kjellén, L.; Welinder, C.; Bengzon, J.; et al. Metastasis Stimulation by Hypoxia and Acidosis-Induced Extracellular Lipid Uptake Is Mediated by Proteoglycan-Dependent Endocytosis. Cancer Res. 2016, 76, 4828–4840. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Chen, J.; Cui, L.; Lu, S.; Xu, S. Amino Acid Metabolism in Tumor Biology and Therapy. Cell Death Dis. 2024, 15, 42. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New Aspects of Amino Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2021, 8, 603837. [Google Scholar] [CrossRef]
- Sharif, T.; Dai, C.; Martell, E.; Ghassemi-Rad, M.S.; Hanes, M.R.; Murphy, P.J.; Kennedy, B.E.; Venugopal, C.; Subapanditha, M.; Giacomantonio, C.A.; et al. TAp73 Modifies Metabolism and Positively Regulates Growth of Cancer Stem–Like Cells in a Redox-Sensitive Manner. Clin. Cancer Res. 2019, 25, 2001–2017. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Ly, S.; Nguyen, K.; Anand, V.; Yuan, B.; El-Dana, F.; Yan, Y.; Arvanitis, Z.; Piyarathna, D.W.B.; Putluri, N.; et al. Metabolic Stress Induces GD2+ Cancer Stem Cell-like Phenotype in Triple-Negative Breast Cancer. Br. J. Cancer 2022, 126, 615–627. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-Driven Glutamine Catabolism Contributes to Prostate Cancer Radiosensitivity by Regulating the Redox State, Stemness and ATG5-Mediated Autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT Inhibition Depletes CD44v-Expressing Tumor Cells That Are Resistant to EGFR-Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.-Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 Identifies Breast Cancer Stem Cells and Promotes Tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, F.; Leonardi, A.; Crescenzi, E. Glutamine Metabolism in Cancer Stem Cells: A Complex Liaison in the Tumor Microenvironment. Int. J. Mol. Sci. 2023, 24, 2337. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fu, Z.; Chen, R.; Zhao, X.; Zhou, Y.; Zeng, B.; Yu, M.; Zhou, Q.; Lin, Q.; Gao, W.; et al. Inhibition of Glutamine Metabolism Counteracts Pancreatic Cancer Stem Cell Features and Sensitizes Cells to Radiotherapy. Oncotarget 2015, 6, 31151–31163. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Nóbrega-Pereira, S. NADPH: New Oxygen for the ROS Theory of Aging. Oncotarget 2016, 7, 50814–50815. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.-P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of Stem-like Cancer Cells by Glutamine through β-Catenin Pathway Mediated by Redox Signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef]
- Prasad, P.; Ghosh, S.; Roy, S.S. Glutamine Deficiency Promotes Stemness and Chemoresistance in Tumor Cells through DRP1-Induced Mitochondrial Fragmentation. Cell. Mol. Life Sci. 2021, 78, 4821–4845. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef]
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the TCA Cycle: Signaling to Tumor Formation. Trends Mol. Med. 2011, 17, 641–649. [Google Scholar] [CrossRef]
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting Glutamine Metabolism as a Therapeutic Strategy for Cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and One-Carbon Metabolism in Cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Cai, H.; Huang, H. Serine Metabolism in Tumor Progression and Immunotherapy. Discov. Oncol. 2025, 16, 628. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Bao, S.; Cai, L.; Wang, M.; Liu, Y.; Sun, Y.; Hu, X. The Role and Research Progress of Serine Metabolism in Tumor Cells. Front. Oncol. 2025, 15, 1509662. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, V.; Jia, W.; Chen, W.-L.; Candi, E.; Melino, G. The Methionine Cycle and Its Cancer Implications. Oncogene 2024, 43, 3483–3488. [Google Scholar] [CrossRef]
- Lim, E.W.; Metallo, C.M. Tracing the Diverse Paths of One-Carbon Metabolism in Cancer and Beyond. Cold Spring Harb. Perspect. Med. 2024, 14, a041533. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The Importance of Serine Metabolism in Cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Cai, Z.; Li, W.; Hager, S.; Wilson, J.L.; Afjehi-Sadat, L.; Heiss, E.H.; Weichhart, T.; Heffeter, P.; Weckwerth, W. Targeting PHGDH Reverses the Immunosuppressive Phenotype of Tumor-Associated Macrophages through α-Ketoglutarate and mTORC1 Signaling. Cell. Mol. Immunol. 2024, 21, 448–465. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; van den Broek, N.J.F.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D.K. Serine, but Not Glycine, Supports One-Carbon Metabolism and Proliferation of Cancer Cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef]
- Subramani, S. A Mammalian Pexophagy Target. Nat. Cell Biol. 2015, 17, 1371–1373. [Google Scholar] [CrossRef]
- Zhao, M.; Hou, Y.; Du, Y.; Yang, L.; Qin, Y.; Peng, M.; Liu, S.; Wan, X.; Qiao, Y.; Zeng, H.; et al. Drosha-Independent miR-6778–5p Strengthens Gastric Cancer Stem Cell Stemness via Regulation of Cytosolic One-Carbon Folate Metabolism. Cancer Lett. 2020, 478, 8–21. [Google Scholar] [CrossRef]
- Qi, Y.-N.; Liu, Z.; Hong, L.-L.; Li, P.; Ling, Z.-Q. Methyltransferase-like Proteins in Cancer Biology and Potential Therapeutic Targeting. J. Hematol. Oncol. 2023, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine Decarboxylase Activity Drives Non-Small Cell Lung Cancer Tumor-Initiating Cells and Tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Endress, J.; Cho, S.; Li, Z.; Zheng, Y.; Asara, J.M.; Blenis, J. Suppression of Nuclear GSK3 Signaling Promotes Serine/One-Carbon Metabolism and Confers Metabolic Vulnerability in Lung Cancer Cells. Sci. Adv. 2022, 8, eabm8786. [Google Scholar] [CrossRef] [PubMed]
- Nigdelioglu, R.; Hamanaka, R.B.; Meliton, A.Y.; O’Leary, E.; Witt, L.J.; Cho, T.; Sun, K.; Bonham, C.; Wu, D.; Woods, P.S.; et al. Transforming Growth Factor (TGF)-β Promotes de Novo Serine Synthesis for Collagen Production. J. Biol. Chem. 2016, 291, 27239–27251. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W.-M. Regulation of Mammalian Nucleotide Metabolism and Biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef]
- Wang, L.; Yang, R.; Kong, Y.; Zhou, J.; Chen, Y.; Li, R.; Chen, C.; Tang, X.; Chen, X.; Xia, J.; et al. Integrative Single-Cell and Bulk Transcriptomes Analyses Reveals Heterogeneity of Serine-Glycine-One-Carbon Metabolism with Distinct Prognoses and Therapeutic Vulnerabilities in HNSCC. Int. J. Oral Sci. 2024, 16, 44. [Google Scholar] [CrossRef]
- Zhang, F.; Ye, J.; Guo, W.; Zhang, F.; Wang, L.; Han, A. TYMS-TM4SF4 Axis Promotes the Progression of Colorectal Cancer by EMT and Upregulating Stem Cell Marker. Am. J. Cancer Res. 2022, 13, 1009. [Google Scholar]
- Lv, Y.; Wang, X.; Li, X.; Xu, G.; Bai, Y.; Wu, J.; Piao, Y.; Shi, Y.; Xiang, R.; Wang, L. Nucleotide de Novo Synthesis Increases Breast Cancer Stemness and Metastasis via cGMP-PKG-MAPK Signaling Pathway. PLoS Biol. 2020, 18, e3000872. [Google Scholar] [CrossRef]
- Liang, T.; Tao, T.; Wu, K.; Liu, L.; Xu, W.; Zhou, D.; Fang, H.; Ding, Q.; Huang, G.; Wu, S. Cancer-Associated Fibroblast-Induced Remodeling of Tumor Microenvironment in Recurrent Bladder Cancer. Adv. Sci. 2023, 10, 2303230. [Google Scholar] [CrossRef]
- Missiaen, R.; Lesner, N.P.; Simon, M.C. HIF: A Master Regulator of Nutrient Availability and Metabolic Cross-talk in the Tumor Microenvironment. EMBO J. 2023, 42, e112067. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic Reprogramming and Crosstalk of Cancer-Related Fibroblasts and Immune Cells in the Tumor Microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [PubMed]
- Cannino, G.; Ciscato, F.; Masgras, I.; Sánchez-Martín, C.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the Cancer-Metabolism Link—Ten Years After. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef]
- Grossi, V.; Lucarelli, G.; Forte, G.; Peserico, A.; Matrone, A.; Germani, A.; Rutigliano, M.; Stella, A.; Bagnulo, R.; Loconte, D.; et al. Loss of STK11 Expression Is an Early Event in Prostate Carcinogenesis and Predicts Therapeutic Response to Targeted Therapy against MAPK/P38. Autophagy 2015, 11, 2102–2113. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Morton, J.D.; Shimomura, L. Sterol Regulatory Element-Binding Proteins: Activators of Cholesterol and Fatty Acid Biosynthesis. Curr. Opin. Lipidol. 1999, 10, 143–150. [Google Scholar] [CrossRef]
- He, Y.; Qi, S.; Chen, L.; Zhu, J.; Liang, L.; Chen, X.; Zhang, H.; Zhuo, L.; Zhao, S.; Liu, S.; et al. The Roles and Mechanisms of SREBP1 in Cancer Development and Drug Response. Genes Dis. 2024, 11, 100987. [Google Scholar] [CrossRef]
- Min, J.-Y.; Kim, D.-H. Stearoyl-CoA Desaturase 1 as a Therapeutic Biomarker: Focusing on Cancer Stem Cells. Int. J. Mol. Sci. 2023, 24, 8951. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Coleman, C.; Sen, T. Stearoyl Coenzyme A Desaturase-1: Multitasker in Cancer, Metabolism, and Ferroptosis. Trends Cancer 2023, 9, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, F.; De Vitis, C.; Maugeri-Saccà, M.; Napoli, C.; Ciliberto, G.; Mancini, R. SCD1, Autophagy and Cancer: Implications for Therapy. J. Exp. Clin. Cancer Res. CR 2021, 40, 265. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef]
- Seitz, R.; Tümen, D.; Kunst, C.; Heumann, P.; Schmid, S.; Kandulski, A.; Müller, M.; Gülow, K. Exploring the Thioredoxin System as a Therapeutic Target in Cancer: Mechanisms and Implications. Antioxidants 2024, 13, 1078. [Google Scholar] [CrossRef]
- Abdullah, N.A.; Md Hashim, N.F.; Muhamad Zakuan, N.; Chua, J.X. Thioredoxin System in Colorectal Cancer: Its Role in Carcinogenesis, Disease Progression, and Response to Treatment. Life Sci. 2024, 348, 122711. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS Homeostasis and Metabolism: A Dangerous Liason in Cancer Cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant Mechanisms and ROS-Related MicroRNAs in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef]
- Fiorillo, M.; Tóth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells 2020, 9, 1529. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The Metabolic Flexibility of Quiescent CSC: Implications for Chemotherapy Resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone Variants—Ancient Wrap Artists of the Epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Radpour, R.; Barekati, Z.; Kohler, C.; Holzgreve, W.; Zhong, X.Y. New Trends in Molecular Biomarker Discovery for Breast Cancer. Genet. Test. Mol. Biomark. 2009, 13, 565–571. [Google Scholar] [CrossRef]
- Lønning, P.E.; Nikolaienko, O.; Knappskog, S. Constitutional Epimutations: From Rare Events Toward Major Cancer Risk Factors? JCO Precis. Oncol. 2025, 9, e2400746. [Google Scholar] [CrossRef]
- Zhou, W.; Reizel, Y. On Correlative and Causal Links of Replicative Epimutations. Trends Genet. TIG 2025, 41, 60–75. [Google Scholar] [CrossRef]
- Mohammad, A.; Jha, S. Epimutations and Their Effect on Chromatin Organization: Exciting Avenues for Cancer Treatment. Cancers 2022, 15, 215. [Google Scholar] [CrossRef]
- Mandhair, H.K.; Novak, U.; Radpour, R. Epigenetic Regulation of Autophagy: A Key Modification in Cancer Cells and Cancer Stem Cells. World J. Stem Cells 2021, 13, 542–567. [Google Scholar] [CrossRef]
- Radpour, R.; Haghighi, M.M.; Fan, A.X.-C.; Torbati, P.M.; Hahn, S.; Holzgreve, W.; Zhong, X.Y. High-Throughput Hacking of the Methylation Patterns in Breast Cancer by In Vitro Transcription and Thymidine-Specific Cleavage Mass Array on MALDI-TOF Silico-Chip. Mol. Cancer Res. 2008, 6, 1702–1709. [Google Scholar] [CrossRef]
- Radpour, R.; Sikora, M.; Grussenmeyer, T.; Kohler, C.; Barekati, Z.; Holzgreve, W.; Lefkovits, I.; Zhong, X.Y. Simultaneous Isolation of DNA, RNA, and Proteins for Genetic, Epigenetic, Transcriptomic, and Proteomic Analysis. J. Proteome Res. 2009, 8, 5264–5274. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, D.; Plass, C. Epigenetic Signatures in Cancer: Proper Controls, Current Challenges and the Potential for Clinical Translation. Genome Med. 2021, 13, 23. [Google Scholar] [CrossRef]
- Radpour, R.; Stucki, M.; Riether, C.; Ochsenbein, A.F. Epigenetic Silencing of Immune-Checkpoint Receptors in Bone Marrow- Infiltrating T Cells in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 663406. [Google Scholar] [CrossRef]
- Esteller, M. Non-Coding RNAs in Human Disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Jaggi, B.; Poon, S.S.S.; Macaulay, C.; Palcic, B. Imaging System for Morphometric Assessment of Absorption or Fluorescence in Stained Cells. Cytometry 1988, 9, 566–572. [Google Scholar] [CrossRef]
- Zhang, P.; Brinton, L.T.; Williams, K.; Sher, S.; Orwick, S.; Tzung-Huei, L.; Mims, A.S.; Coss, C.C.; Kulp, S.K.; Youssef, Y.; et al. Targeting DNA Damage Repair Functions of Two Histone Deacetylases, HDAC8 and SIRT6, Sensitizes Acute Myeloid Leukemia to NAMPT Inhibition. Clin. Cancer Res. 2021, 27, 2352–2366. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Liu, C.; Yan, K.; Wang, X.; Sheng, X. Interactions between Long Non-Coding RNAs and M6 A Modification in Cancer. Discov. Oncol. 2025, 16, 579. [Google Scholar] [CrossRef]
- Peng, J.; Liu, W.; Tian, J.; Shu, Y.; Zhao, R.; Wang, Y. Non-Coding RNAs as Key Regulators of Epithelial-Mesenchymal Transition in Breast Cancer. Front. Cell Dev. Biol. 2025, 13, 1544310. [Google Scholar] [CrossRef]
- Solaimani, M.; Hosseinzadeh, S.; Abasi, M. Non-Coding RNAs, a Double-Edged Sword in Breast Cancer Prognosis. Cancer Cell Int. 2025, 25, 123. [Google Scholar] [CrossRef]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the Modules of EMT and Stemness: A Tunable ‘Stemness Window’ Model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT Programs Control Normal Mammary Stem Cells and Tumour-Initiating Cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-Associated Fibroblasts: The Chief Architect in the Tumor Microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef]
- Di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the Kynurenine Pathway Predicts Poor Outcome in Patients with Clear Cell Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 461.e15–461.e27. [Google Scholar] [CrossRef]
- Qian, J.; Rankin, E.B. Hypoxia-Induced Phenotypes That Mediate Tumor Heterogeneity. In Hypoxia and Cancer Metastasis; Gilkes, D.M., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1136, pp. 43–55. ISBN 978-3-030-12733-6. [Google Scholar]
- De Francesco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting Hypoxic Cancer Stem Cells (CSCs) with Doxycycline: Implications for Optimizing Anti-Angiogenic Therapy. Oncotarget 2017, 8, 56126–56142. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Peiris-Pages, M.; Ozsvari, B.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Targeting Cancer Stem Cell Propagation with Palbociclib, a CDK4/6 Inhibitor: Telomerase Drives Tumor Cell Heterogeneity. Oncotarget 2017, 8, 9868–9884. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective Mitochondrial Transfer from Bone Marrow Stromal Cells to Acute Myeloid Leukemic Cells during Chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Ye, X.-Q.; Wang, G.-H.; Huang, G.-J.; Bian, X.-W.; Qian, G.-S.; Yu, S.-C. Heterogeneity of Mitochondrial Membrane Potential: A Novel Tool to Isolate and Identify Cancer Stem Cells from a Tumor Mass? Stem Cell Rev. Rep. 2011, 7, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; De Francesco, E.M.; De Boer, R.; Tanowitz, H.B.; Lisanti, M.P. NADH Autofluorescence, a New Metabolic Biomarker for Cancer Stem Cells: Identification of Vitamin C and CAPE as Natural Products Targeting “Stemness”. Oncotarget 2017, 8, 20667–20678. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin Inhibits Cell Proliferation, Migration and Invasion by Attenuating CSC Function Mediated by Deregulating miRNAs in Pancreatic Cancer Cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II Clinical Trial of Metformin as a Cancer Stem Cell-Targeting Agent in Ovarian Cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Kaur, G.; Sharma, P.; Dogra, N.; Singh, S. Eradicating Cancer Stem Cells: Concepts, Issues, and Challenges. Curr. Treat. Options Oncol. 2018, 19, 20. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain Tumor Initiating Cells Adapt to Restricted Nutrition through Preferential Glucose Uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Lucarelli, G.; Lasorsa, F.; Milella, M.; d’Amati, A.; Ingravallo, G.; Silecchia, M.; Errede, M.; Bianchi, C.; Spilotros, M.; Battaglia, M.; et al. Transcriptomic and Proteo-Metabolic Determinants of the Grading System in Clear Cell Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2025; in press. [Google Scholar] [CrossRef]

{kind=link}
| Therapeutic Strategy | Drug | Clinical Trials |
|---|---|---|
| Glycolysis | 2-DG Lonidamine | NCT00633087 NCT02758860 |
| OXPHOS | Metformin Oligomycin Complex I inhibitors | NCT01579812 none NCT02882321, NCT03291938 |
| Glutaminolysis | CB-839 (glutaminase inhibitor) | NCT02071862, NCT03163667, NCT03872427 |
| Lipogenesis | Orlistat (FASN inhibitor) | none |
| Fatty acid β-oxidation | Etomoxir (CPT1 inhibitor) | none |
| ROS production | Salinomycin Fenretinide | none NCT00646230 |
| mTOR | Sirolimus | NCT03433183 |
| Everolimus | NCT00876395, NCT01007942, | |
| NCT04485559 | ||
| Temsirolimus | NCT01614197, NCT03433183 | |
| Ridaforolimus | NCT00538239 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milella, M.; Rutigliano, M.; Pandolfo, S.D.; Aveta, A.; Crocetto, F.; Ferro, M.; d’Amati, A.; Ditonno, P.; Lucarelli, G.; Lasorsa, F. The Metabolic Landscape of Cancer Stem Cells: Insights and Implications for Therapy. Cells 2025, 14, 717. https://doi.org/10.3390/cells14100717
Milella M, Rutigliano M, Pandolfo SD, Aveta A, Crocetto F, Ferro M, d’Amati A, Ditonno P, Lucarelli G, Lasorsa F. The Metabolic Landscape of Cancer Stem Cells: Insights and Implications for Therapy. Cells. 2025; 14(10):717. https://doi.org/10.3390/cells14100717
Chicago/Turabian StyleMilella, Martina, Monica Rutigliano, Savio Domenico Pandolfo, Achille Aveta, Felice Crocetto, Matteo Ferro, Antonio d’Amati, Pasquale Ditonno, Giuseppe Lucarelli, and Francesco Lasorsa. 2025. "The Metabolic Landscape of Cancer Stem Cells: Insights and Implications for Therapy" Cells 14, no. 10: 717. https://doi.org/10.3390/cells14100717
APA StyleMilella, M., Rutigliano, M., Pandolfo, S. D., Aveta, A., Crocetto, F., Ferro, M., d’Amati, A., Ditonno, P., Lucarelli, G., & Lasorsa, F. (2025). The Metabolic Landscape of Cancer Stem Cells: Insights and Implications for Therapy. Cells, 14(10), 717. https://doi.org/10.3390/cells14100717