

Genomic and Phenotypic Characterization of CHO 4BGD Cells with Quad Knockout and Overexpression of Two Housekeeping Genes That Allow for Metabolic Selection and Extended Fed-Batch Culturing
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. NGS Analysis of CHO 4BGD Cells
3.2. Analysis of Potential Off-Target DNA Editing Events
3.3. Analysis of Genome-Integrated Plasmids and Expression of Plasmid-Encoded Proteins
3.4. Phenotype of 4BGD Cells
3.5. Fed-Batch Culturing of 4BGD -Derived Cell Line
3.6. Macroautophagy Markers Dynamics in 4BGD-mAb Cell Line
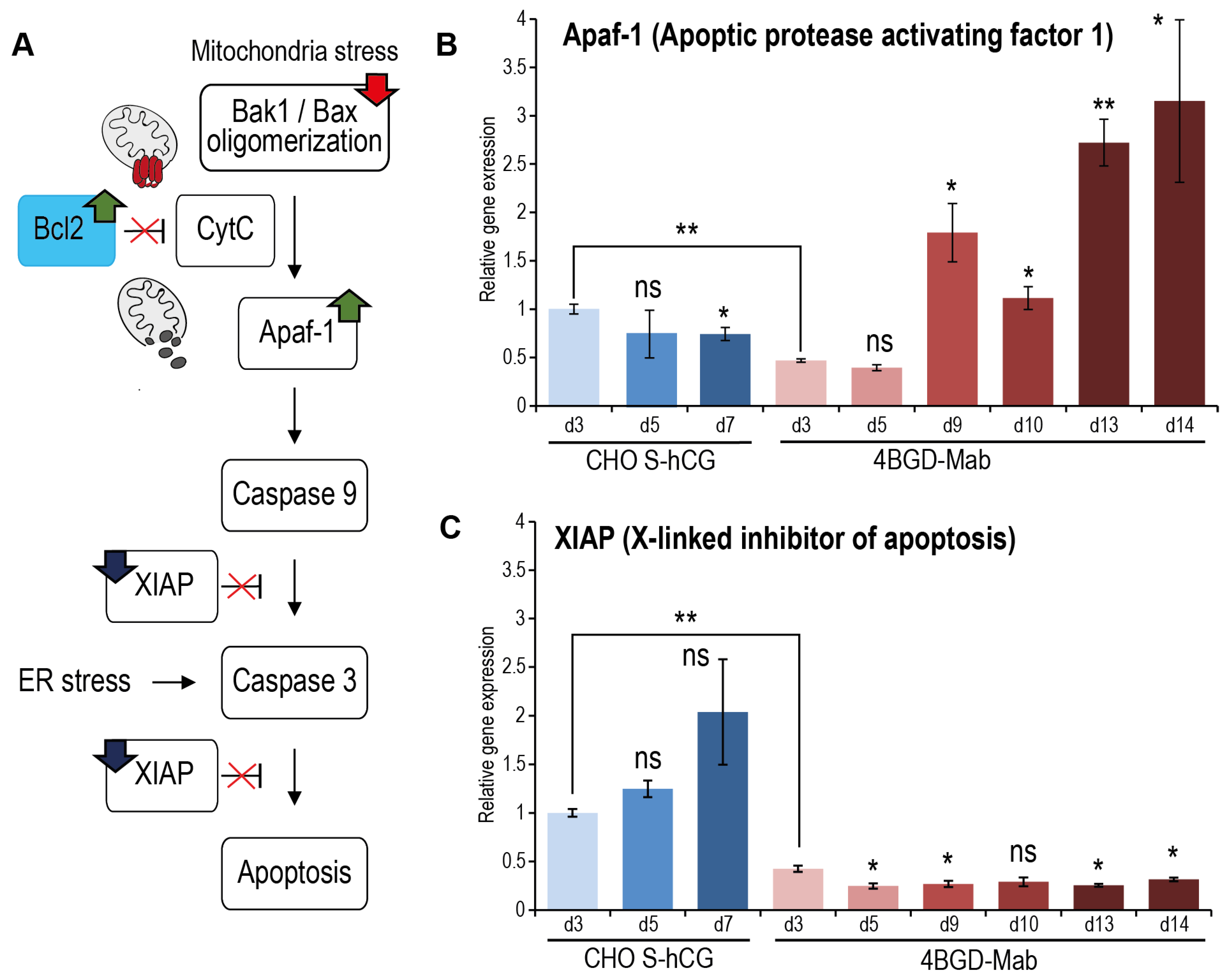
3.7. qPCR Analysis of Genes Involved in Apoptosis and Macroautophagy Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CHO | Chinese Hamster Ovary (cells) |
| CRISPR/Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 |
| DHFR | Dihydrofolate Reductase (selection marker gene) |
| GS | Glutamine Synthetase (selection marker gene) |
| Bak1 | BCL2 Antagonist/Killer 1 (pro-apoptotic gene) |
| Bax | BCL2-Associated X Protein (pro-apoptotic gene) |
| Bcl-2 | B-cell Lymphoma 2 (anti-apoptotic gene) |
| Beclin-1 | Autophagy-related protein (enhances macroautophagy) |
| NGS | Next-Generation Sequencing (high-throughput DNA sequencing) |
| gRNA | Guide RNA (used in CRISPR/Cas9 for targeting specific DNA sequences) |
| MTX | Methotrexate (DHFR inhibitor used for gene amplification) |
| ELISA | Enzyme-Linked Immunosorbent Assay (protein detection method) |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction (gene expression analysis) |
| qPCR | Quantitative Polymerase Chain Reaction (quantitative gene expression analysis) |
| Fc | Fragment crystallizable (region of an antibody) |
| IgG | Immunoglobulin G (antibody type) |
| mAb | Monoclonal Antibody |
| EEF1A1 | Eukaryotic Translation Elongation Factor 1 Alpha 1 (promoter for gene expression) |
| PAM | Protospacer Adjacent Motif (CRISPR/Cas9 target sequence requirement) |
| ORF | Open Reading Frame (coding sequence of a gene) |
| SNP | Single Nucleotide Polymorphism (genetic variation) |
| Indel | Insertion/Deletion (genetic mutation) |
| CDS | Coding DNA Sequence (region of DNA that codes for a protein) |
| LC3 | Microtubule-associated protein light chain 3 (autophagy marker) |
| Apaf-1 | Apoptotic Protease Activating Factor 1 (apoptosis-related protein) |
| XIAP | X-linked Inhibitor of Apoptosis Protein (apoptosis inhibitor) |
| ULK1 | Unc-51 Like Autophagy Activating Kinase 1 (autophagy-related kinase) |
| ATG | Autophagy-related Gene (genes involved in autophagy) |
| VPS34 | Vacuolar Protein Sorting 34 (autophagy-related kinase) |
| AMPK | AMP-activated Protein Kinase (energy-sensing enzyme) |
| mTOR | Mechanistic Target of Rapamycin (regulates cell growth and autophagy) |
| FIP200 | Focal Adhesion Kinase Family Interacting Protein of 200 kD (autophagy-related protein) |
| Ambra1 | Autophagy and Beclin-1 Regulator 1 (autophagy-related protein) |
| PE | Phosphatidylethanolamine (lipid involved in autophagy) |
| PAR | Poly(ADP-ribose) (marker of parthanatos, a form of cell death) |
| FSP1 | Ferroptosis Suppressor Protein 1 (marker of ferroptosis, a form of cell death) |
| MDA | Malondialdehyde (marker of lipid peroxidation in ferroptosis) |
| GPX4 | Glutathione Peroxidase 4 (enzyme that prevents ferroptosis) |
| ER | Endoplasmic Reticulum |
References
- Turilova, V.I.; Goryachaya, T.S.; Yakovleva, T.K. Chinese hamster ovary cell line DXB-11: Chromosomal instability and karyotype heterogeneity. Mol. Cytogenet. 2021, 14, 11. [Google Scholar] [CrossRef]
- Derouazi, M.; Martinet, D.; Besuchet Schmutz, N.; Flaction, R.; Wicht, M.; Bertschinger, M.; Hacker, D.L.; Beckmann, J.S.; Wurm, F.M. Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem. Biophys. Res. Commun. 2006, 340, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Boch, J. TALE and TALEN genome editing technologies. Gene Genome Ed. 2021, 2, 100007. [Google Scholar] [CrossRef]
- Malphettes, L.; Freyvert, Y.; Chang, J.; Liu, P.Q.; Chan, E.; Miller, J.C.; Zhou, Z.; Nguyen, T.; Tsai, C.; Snowden, A.W.; et al. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol. Bioeng. 2010, 106, 774–783. [Google Scholar] [CrossRef]
- Liu, W.; Padmashali, R.; Monzon, O.Q.; Lundberg, D.; Jin, S.; Dwyer, B.; Lee, Y.J.; Korde, A.; Park, S.; Pan, C.; et al. Generation of FX(-/-) and Gmds(-/-) CHOZN host cell lines for the production of afucosylated therapeutic antibodies. Biotechnol. Prog. 2021, 37, e3061. [Google Scholar] [CrossRef] [PubMed]
- McCarty, N.S.; Graham, A.E.; Studena, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef]
- Ronda, C.; Pedersen, L.E.; Hansen, H.G.; Kallehauge, T.B.; Betenbaugh, M.J.; Nielsen, A.T.; Kildegaard, H.F. Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol. Bioeng. 2014, 111, 1604–1616. [Google Scholar] [CrossRef]
- Henry, M.N.; MacDonald, M.A.; Orellana, C.A.; Gray, P.P.; Gillard, M.; Baker, K.; Nielsen, L.K.; Marcellin, E.; Mahler, S.; Martinez, V.S. Attenuating apoptosis in Chinese hamster ovary cells for improved biopharmaceutical production. Biotechnol. Bioeng. 2020, 117, 1187–1203. [Google Scholar] [CrossRef]
- Cost, G.J.; Freyvert, Y.; Vafiadis, A.; Santiago, Y.; Miller, J.C.; Rebar, E.; Collingwood, T.N.; Snowden, A.; Gregory, P.D. BAK and BAX deletion using zinc-finger nucleases yields apoptosis-resistant CHO cells. Biotechnol. Bioeng. 2010, 105, 330–340. [Google Scholar] [CrossRef]
- MacDonald, M.A.; Barry, C.; Groves, T.; Martinez, V.S.; Gray, P.P.; Baker, K.; Shave, E.; Mahler, S.; Munro, T.; Marcellin, E.; et al. Modeling apoptosis resistance in CHO cells with CRISPR-mediated knockouts of Bak1, Bax, and Bok. Biotechnol. Bioeng. 2022, 119, 1380–1391. [Google Scholar] [CrossRef]
- Tang, D.; Lam, C.; Bauer, N.; Auslaender, S.; Snedecor, B.; Laird, M.W.; Misaghi, S. Bax and Bak knockout apoptosis-resistant Chinese hamster ovary cell lines significantly improve culture viability and titer in intensified fed-batch culture process. Biotechnol. Prog. 2022, 38, e3228. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.A.; Nobel, M.; Martinez, V.S.; Baker, K.; Shave, E.; Gray, P.P.; Mahler, S.; Munro, T.; Nielsen, L.K.; Marcellin, E. Engineering death resistance in CHO cells for improved perfusion culture. MAbs 2022, 14, 2083465. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Ha, T.K.; Park, J.H.; Lee, G.M. Anti-cell death engineering of CHO cells: Co-overexpression of Bcl-2 for apoptosis inhibition, Beclin-1 for autophagy induction. Biotechnol. Bioeng. 2013, 110, 2195–2207. [Google Scholar] [CrossRef]
- Orlova, N.A.; Dayanova, L.K.; Gayamova, E.A.; Sinegubova, M.V.; Kovnir, S.V.; Vorobiev, I.I. Targeted Knockout of the dhfr, glul, bak1, and bax Genes by the Multiplex Genome Editing in CHO Cells. Dokl. Biochem. Biophys. 2022, 502, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Kovnir, S.W.; Dayanova, L.K.; Gayamova, E.A.; Dybovsky, L.N.; Vorobiev, I.I.; Orlova, N.A. Knockout of bax, bak1 genes and overexpression of bcl2, becn1 genes increase lifespan and maximum density of CHO-S cell culture. Russ. J. Biotechnol. 2022, 38, 16–22. [Google Scholar] [CrossRef]
- Zischewski, J.; Fischer, R.; Bortesi, L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017, 35, 95–104. [Google Scholar] [CrossRef]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef]
- Slesarev, A.; Viswanathan, L.; Tang, Y.; Borgschulte, T.; Achtien, K.; Razafsky, D.; Onions, D.; Chang, A.; Cote, C. CRISPR/CAS9 targeted CAPTURE of mammalian genomic regions for characterization by NGS. Sci. Rep. 2019, 9, 3587. [Google Scholar] [CrossRef]
- Orlova, N.A.; Kovnir, S.V.; Hodak, J.A.; Vorobiev, I.I.; Gabibov, A.G.; Skryabin, K.G. Improved elongation factor-1 alpha-based vectors for stable high-level expression of heterologous proteins in Chinese hamster ovary cells. BMC Biotechnol. 2014, 14, 56. [Google Scholar] [CrossRef]
- Ryazanova, A.Y.; Khodak, Y.A.; Orlova, N.A.; Sinegubova, M.V.; Dayanova, L.K.; Kovnir, S.V.; Korobova, S.V.; Ledov, V.A.; Kovalchuk, A.L.; Alkhazova, B.I.; et al. Receptor-binding domain of SARS-CoV-2 spike protein fused to a non-glycosylated crystallizable fragment of human IgG1: Obtaining and assessment of immunogenicity. Russ. J. Biotechnol. 2022, 38, 12–19. [Google Scholar] [CrossRef]
- Smith, T.J.; Lou, J.; Geren, I.N.; Forsyth, C.M.; Tsai, R.; Laporte, S.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Smith, L.A.; et al. Sequence variation within botulinum neurotoxin serotypes impacts antibody binding and neutralization. Infect. Immun. 2005, 73, 5450–5457. [Google Scholar] [CrossRef] [PubMed]
- Orlova, N.A.; Kovnir, S.V.; Khodak, Y.A.; Polzikov, M.A.; Nikitina, V.A.; Skryabin, K.G.; Vorobiev, I.I. High-level expression of biologically active human follicle stimulating hormone in the Chinese hamster ovary cell line by a pair of tricistronic and monocistronic vectors. PLoS ONE 2019, 14, e0219434. [Google Scholar] [CrossRef]
- Brown, A.J.; Gibson, S.; Hatton, D.; James, D.C. Transcriptome-Based Identification of the Optimal Reference CHO Genes for Normalisation of qPCR Data. Biotechnol. J. 2018, 13, 1700259. [Google Scholar] [CrossRef] [PubMed]
- Tournayre, J.; Reichstadt, M.; Parry, L.; Fafournoux, P.; Jousse, C. “Do my qPCR calculation”, a web tool. Bioinformation 2019, 15, 369–372. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Brier, L.W.; Ge, L.; Stjepanovic, G.; Thelen, A.M.; Hurley, J.H.; Schekman, R. Regulation of LC3 lipidation by the autophagy-specific class III phosphatidylinositol-3 kinase complex. Mol. Biol. Cell 2019, 30, 1098–1107. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, Y.; Hu, Q.; Bai, X.C.; Huang, W.; Yan, C.; Scheres, S.H.; Shi, Y. Atomic structure of the apoptosome: Mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes. Dev. 2015, 29, 2349–2361. [Google Scholar] [CrossRef]
- Kim, H.E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef]
- Ho, A.T.; Li, Q.H.; Okada, H.; Mak, T.W.; Zacksenhaus, E. XIAP activity dictates Apaf-1 dependency for caspase 9 activation. Mol. Cell Biol. 2007, 27, 5673–5685. [Google Scholar] [CrossRef]
- Bratton, S.B.; Lewis, J.; Butterworth, M.; Duckett, C.S.; Cohen, G.M. XIAP inhibition of caspase-3 preserves its association with the Apaf-1 apoptosome and prevents CD95- and Bax-induced apoptosis. Cell Death Differ. 2002, 9, 881–892. [Google Scholar] [CrossRef]
- Chakraborty, R. Understanding the role of Atg7 and Atg14 in autophagy. Senior Honors Theses and Projects; Eastern Michigan University: Ypsilanti, MI, USA, 2019. [Google Scholar]
- Reinhart, D.; Damjanovic, L.; Kaisermayer, C.; Sommeregger, W.; Gili, A.; Gasselhuber, B.; Castan, A.; Mayrhofer, P.; Grunwald-Gruber, C.; Kunert, R. Bioprocessing of Recombinant CHO-K1, CHO-DG44, and CHO-S: CHO Expression Hosts Favor Either mAb Production or Biomass Synthesis. Biotechnol. J. 2019, 14, e1700686. [Google Scholar] [CrossRef] [PubMed]
- Kaas, C.S.; Kristensen, C.; Betenbaugh, M.J.; Andersen, M.R. Sequencing the CHO DXB11 genome reveals regional variations in genomic stability and haploidy. BMC Genom. 2015, 16, 160. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, S.; Qu, Y.; Snowden, A.; Chang, J.; Snedecor, B. Resilient immortals, characterizing and utilizing Bax/Bak deficient Chinese hamster ovary (CHO) cells for high titer antibody production. Biotechnol. Prog. 2013, 29, 727–737. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Pullaiah, T.; Swamy, M.K.; Chen, Z.-S. Paclitaxel: Sources, Chemistry, Anticancer Actions, and Current Biotechnology, 1st ed.; Elsevier: Waltham, MA, USA, 2022. [Google Scholar]
- Khing, T.M.; Choi, W.S.; Kim, D.M.; Po, W.W.; Thein, W.; Shin, C.Y.; Sohn, U.D. The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells. Sci. Rep. 2021, 11, 23490. [Google Scholar] [CrossRef]
- Mentlak, D.A.; Raven, J.; Moses, T.; Massie, F.; Barber, N.; Hoare, R.; Burton, G.; Young, A.; Pybus, L.P.; Rosser, S.; et al. Dissecting cell death pathways in fed-batch bioreactors. Biotechnol. J. 2024, 19, e2300257. [Google Scholar] [CrossRef] [PubMed]
- Astley, K.; Al-Rubeai, M. The role of Bcl-2 and its combined effect with p21CIP1 in adaptation of CHO cells to suspension and protein-free culture. Appl. Microbiol. Biotechnol. 2008, 78, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Catalán-Tatjer, D.; Ganesan, S.K.; Martínez-Monge, I.; Grav, L.M.; Lavado-García, J.; Nielsen, L.K. Evaluating apoptotic gene efficiency for CHO culture performance using targeted integration. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Pedro, J.M.; Wei, Y.; Sica, V.; Maiuri, M.C.; Zou, Z.; Kroemer, G.; Levine, B. BAX and BAK1 are dispensable for ABT-737-induced dissociation of the BCL2-BECN1 complex and autophagy. Autophagy 2015, 11, 452–459. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Wold, M.S.; Lim, J.; Lachance, V.; Deng, Z.; Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016, 11, 76. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Nucleotide Sequence 5′ → 3′ |
|---|---|
| RT-Mmadhc-F | TGTCACCTCAATGGGACTGC |
| RT-Mmadhc-R | CAGGTGCATCACTACTCTGAAAC |
| RT-bACT-F | GCTCTTTTCCAGCCTTCCTT |
| RT-bACT-R | GAGCCAGAGCAGTGATCTCC |
| RT-XIAP-R | TTTACACCCTGGGTACCACT |
| RT-XIAP-F | GAGGAGGGCTCACAGATTGG |
| RT-APAF1-R | TGTCAAACATGAGCCAAGCC |
| RT-APAF1-F | CTGCCTAAGGCGAGCGATTA |
| RT-Ambra1-F | CAGTCTGCTGTGGCCAGTAA |
| RT-Ambra1-R | TCACGGAGGCATTGCTGATT |
| RT-ATG14-58F | GCTTCTTCTGTTGGTAGACTTG |
| RT-ATG14-58R | CGAGGTGGAGGATATAGAGTTAG |
| RT-ATG7-58F | AAGGCTGGCTGAGTCATC |
| RT-ATG7-58R | TGTCCAAGTCCAAGGTAGG |
| RT-ULK1-58F | CAAGAAGAACCTCGCCAAGTC |
| RT-ULK1-58R | GGAAGTCATACAACGCCACAA |
| TA-DHFR-1v7-F | GGGGACCCTGGTCACGTGTGCT |
| TA-DHFR-1v7-R | GCCTGGTGGTGGAGGCGGAGTCT |
| TA-GS-2v4-F | TCTTCGTGTTTGTCATAAGCC |
| TA-GS-2v4-R | TAACTGGGCACGAGGAATAAA |
| SQ-BAX456-F | TGTCTCCCTCGTAGCCCCTAT |
| SQ-BAX456-R | AGCCTTGCTTGTTTTGTTCG |
| SQ-BAK467-F | CTGTGCTCTTGGTTTCTTTCACG |
| SQ-BAK467-R | AGGGGTGGGTTACAGAGTGGC |
| Gene Name | gRNA Sequence | Number of Potential Off-Target Sites | Off-Target Mutations Found | Potentially Affected Genes |
|---|---|---|---|---|
| Bak1 | GGAAGCCGGTCAAACCACGT | 158 | 2 | ABCC4 |
| Bax | GCTGATGGCAACTTCAACTG | 39 | 3 | CUNH12orf56, Nepro |
| Glul | GGCCTCCTCGATGTGCCTGG | 86 | 2 | Cep162 (heterozygous) |
| DHFR | GCAAGAACGGAGACCTTCCC | 65 | 4 | Sumf1 |
| DHFR | GGTGATCGCTGCTGCTGTCA | 120 | 3 | - |
| DNA Region | ORF | Mean Coverage | Calculated Copy Number per Diploid Genome |
|---|---|---|---|
| CHO genome | - | 9.97527 | 1 |
| p1.2-Zeo-Bcl2 plasmid | Bcl-2 | 5.12919 | 1.0 |
| p1.2-Zeo-Bcl2 plasmid | Zeocin resistance | 8.85867 | 1.8 |
| p1.2-Hygro-Beclin1 plasmid | Beclin1 | 10.3415 | 2.1 |
| p1.2-Hygro-Beclin1 plasmid | Hygromycin resistance | 20.769 | 4.2 |
| Bcl2 (Gene ID: 100774873) | - | 9.76513 | 1.0 |
| Becn1 (Gene ID: 100762045) | - | 9.46119 | 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlova, N.A.; Sinegubova, M.V.; Kolesov, D.E.; Khodak, Y.A.; Tatarskiy, V.V.; Vorobiev, I.I. Genomic and Phenotypic Characterization of CHO 4BGD Cells with Quad Knockout and Overexpression of Two Housekeeping Genes That Allow for Metabolic Selection and Extended Fed-Batch Culturing. Cells 2025, 14, 692. https://doi.org/10.3390/cells14100692
Orlova NA, Sinegubova MV, Kolesov DE, Khodak YA, Tatarskiy VV, Vorobiev II. Genomic and Phenotypic Characterization of CHO 4BGD Cells with Quad Knockout and Overexpression of Two Housekeeping Genes That Allow for Metabolic Selection and Extended Fed-Batch Culturing. Cells. 2025; 14(10):692. https://doi.org/10.3390/cells14100692
Chicago/Turabian StyleOrlova, Nadezhda Alexandrovna, Maria Valerievna Sinegubova, Denis Eduardovich Kolesov, Yulia Alexandrovna Khodak, Victor Vyacheslavovich Tatarskiy, and Ivan Ivanovich Vorobiev. 2025. "Genomic and Phenotypic Characterization of CHO 4BGD Cells with Quad Knockout and Overexpression of Two Housekeeping Genes That Allow for Metabolic Selection and Extended Fed-Batch Culturing" Cells 14, no. 10: 692. https://doi.org/10.3390/cells14100692
APA StyleOrlova, N. A., Sinegubova, M. V., Kolesov, D. E., Khodak, Y. A., Tatarskiy, V. V., & Vorobiev, I. I. (2025). Genomic and Phenotypic Characterization of CHO 4BGD Cells with Quad Knockout and Overexpression of Two Housekeeping Genes That Allow for Metabolic Selection and Extended Fed-Batch Culturing. Cells, 14(10), 692. https://doi.org/10.3390/cells14100692