
Identification of Prognostic Biomarkers of Ovarian High-Grade Serous Carcinoma: A Preliminary Study Using Spatial Transcriptome Analysis and Multispectral Imaging

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Construction of Tissue Microarray (TMA)
2.2. Spatial Transcriptome Profiling
2.3. Spatial Transcriptome Data Processing
2.4. Gene Expression Analysis
2.5. Multispectral Imaging of Immune Cells Using VECTRA
2.6. Integrative Analysis of Multispectral Immune Cell and Spatial Transcriptome Data
2.7. Statistical Analysis
3. Results
3.1. Spatial Transcriptome Analysis and Gene Profiling of Selected ROI
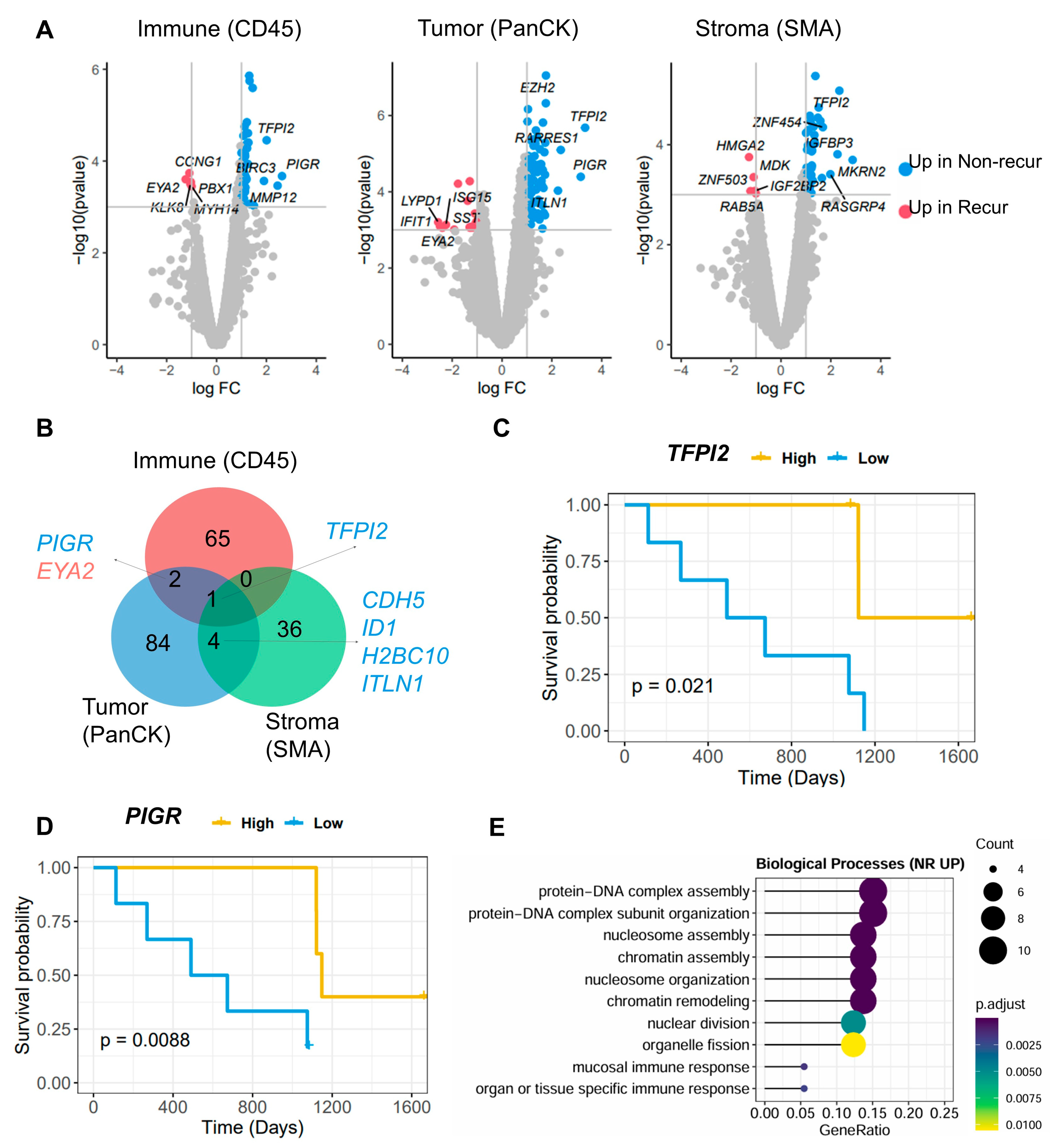
3.2. Identification of Differentially Expressed Genes (DEGs) Between the Non-Recur and Recur Groups
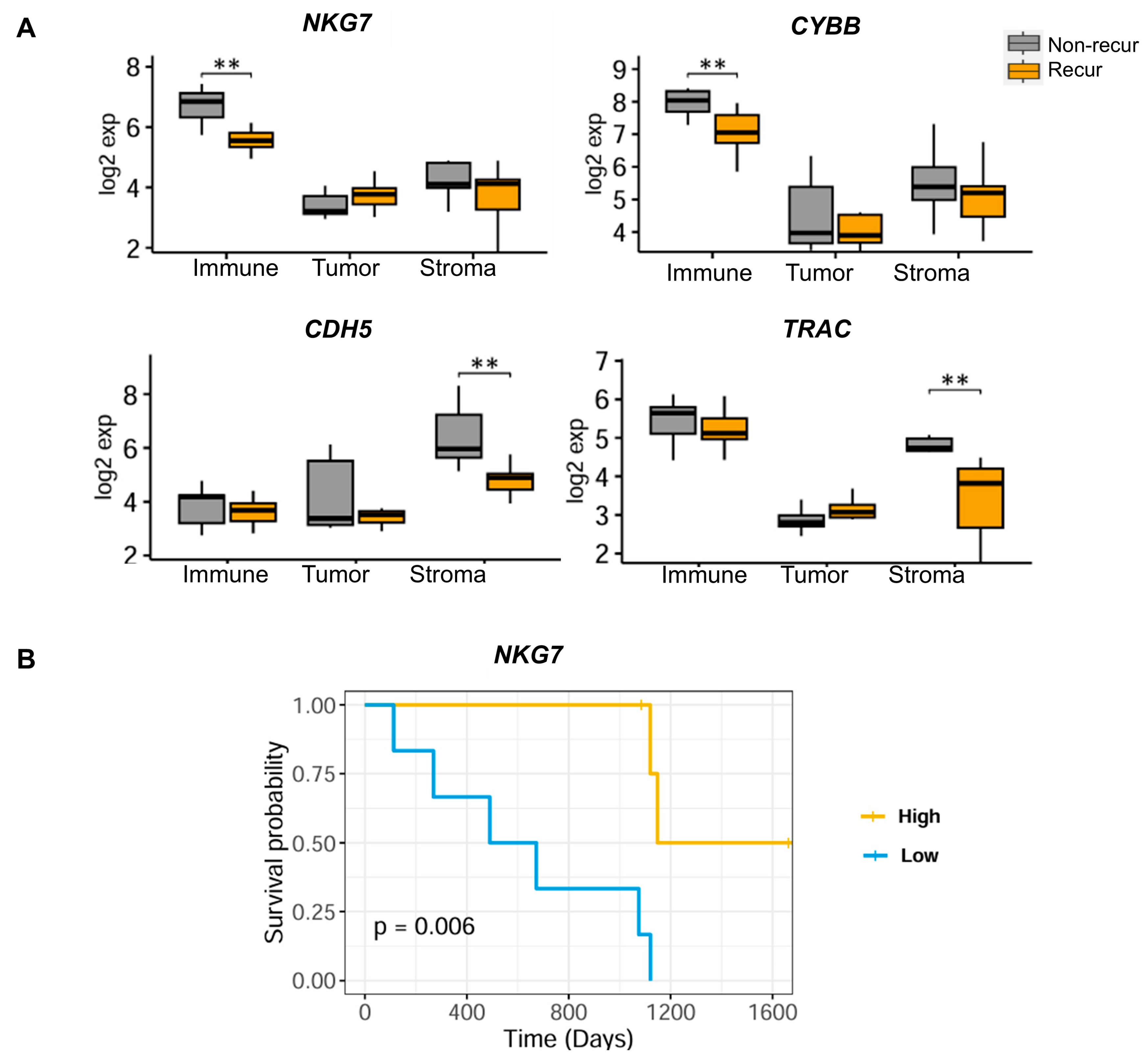
3.3. Identification of TME-Related Markers
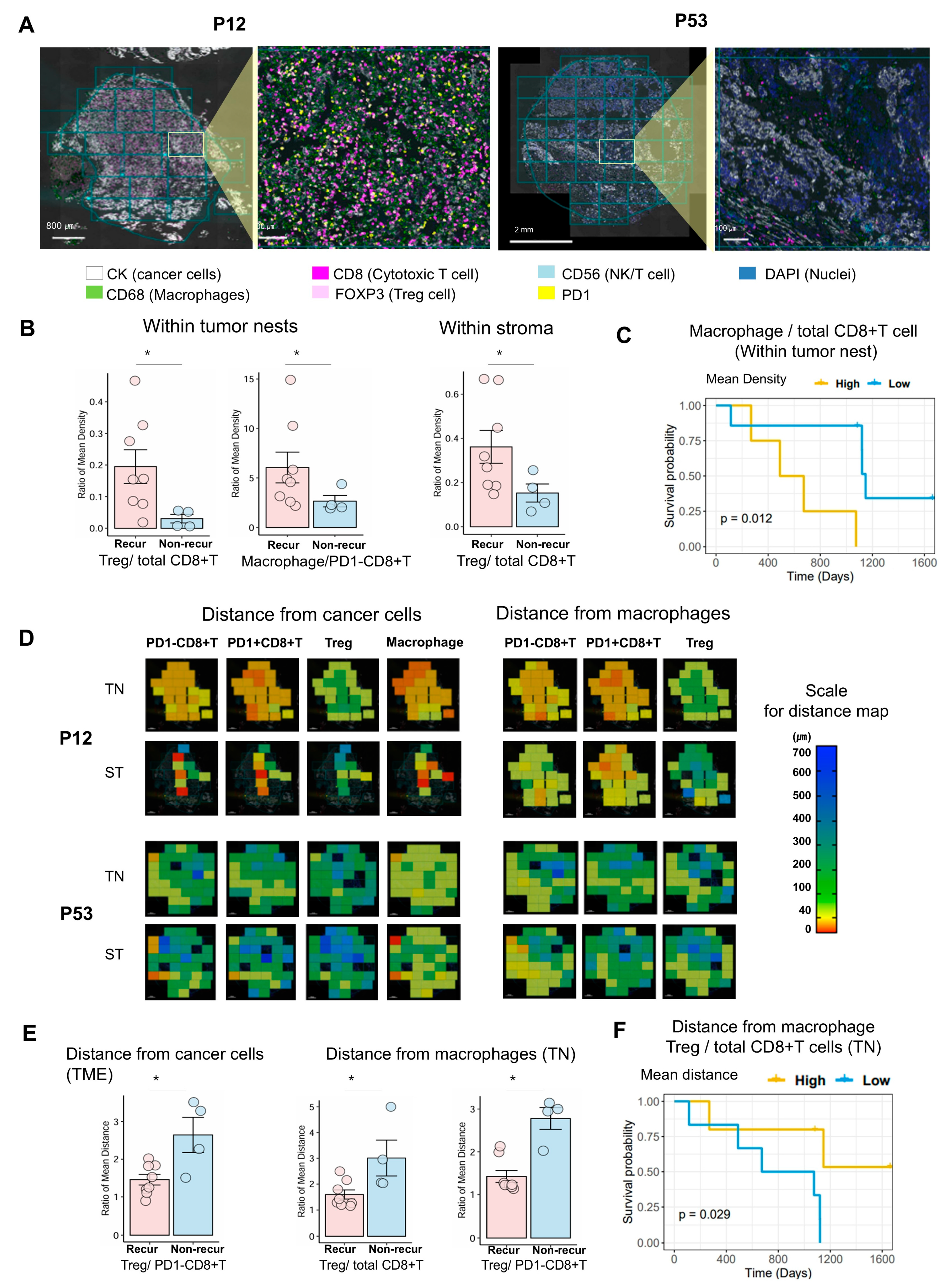
3.4. Multispectral Quantitative Computational Assessment of Immune Cell Density and Distance Between Tumor and Immune Cells Within Tumor Nests and Stroma
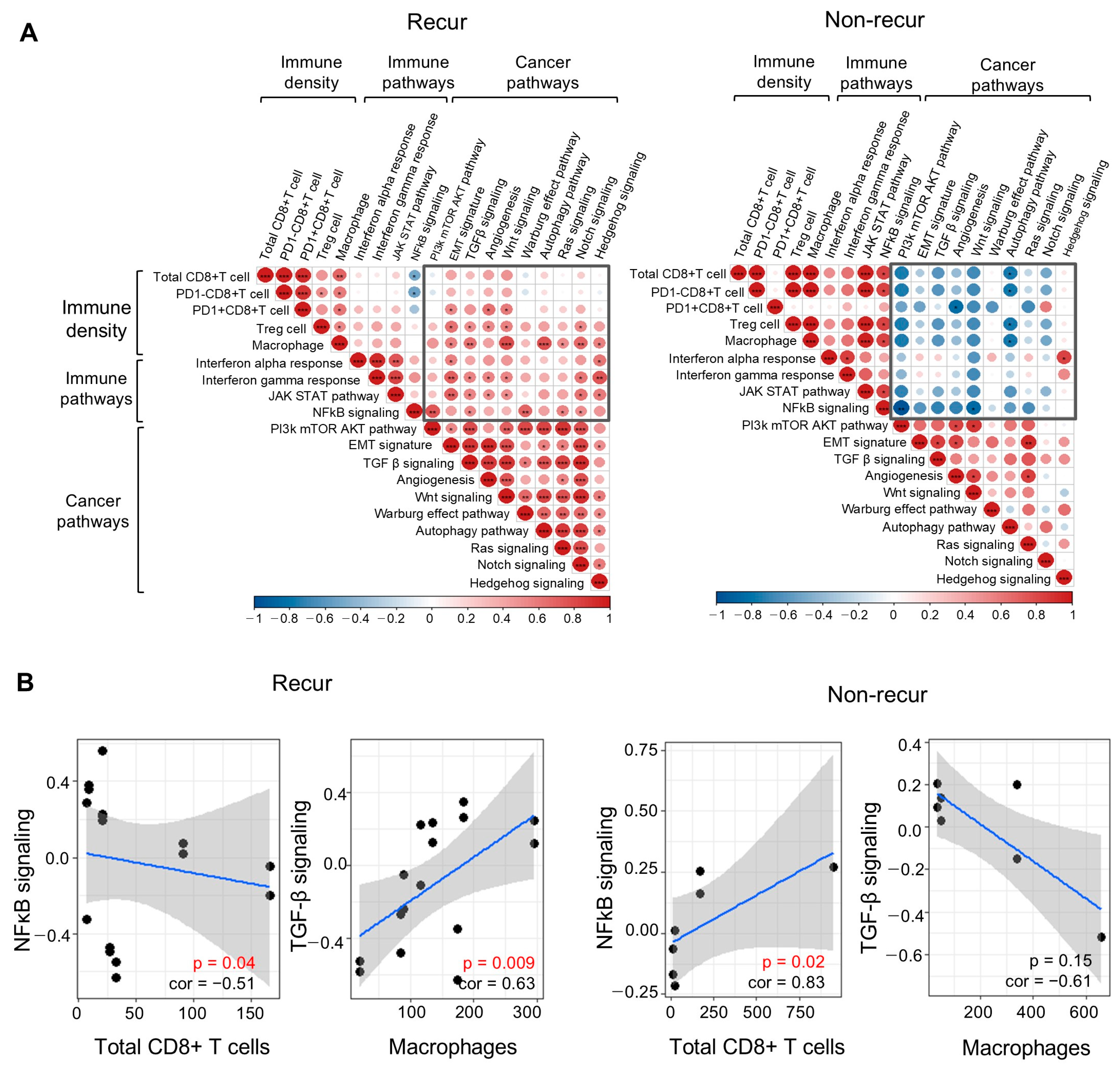
3.5. Integrative Analysis of Multispectral Immune Cell Density and Spatial Transcriptome Data Related to the TME
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| adj. | adjusted |
| AOI | areas of interest |
| CDH5 | cadherin 5 |
| CYBB | cytochrome b-245 beta chain |
| DEGs | differentially expressed genes |
| DSP | Digital Spatial Profiling |
| Dx | diagnosis |
| FC | fold change |
| FFPE | formalin-fixed paraffin-embedded |
| HGSC | high-grade serous carcinomas |
| IF | immunofluorescence |
| LOQ | limit of quantitation |
| NKG7 | natural killer cell granule protein 7 |
| OC | ovarian cancer |
| PCR | Polymerase Chain Reaction |
| PFS | progression-free survival |
| PIGR | polymeric immunoglobulin receptor |
| ROI | regions of interest |
| TFPI2 | tissue factor pathway inhibitor 2 |
| TMA | tissue microarray |
| TME | tumor microenvironment |
| TN | tumor nest |
| TRAC | T cell receptor alpha constant |
| Tregs | regulatory T cells |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Pogge von Strandmann, E.; Reinartz, S.; Wager, U.; Muller, R. Tumor-Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends Cancer 2017, 3, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Zollinger, D.R.; Lingle, S.E.; Sorg, K.; Beechem, J.M.; Merritt, C.R. GeoMx RNA Assay: High Multiplex, Digital, Spatial Analysis of RNA in FFPE Tissue. Methods Mol. Biol. 2020, 2148, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.K.; Brambs, C.E.; Hiller, G.G.R.; May, D.; Schmoeckel, E.; Horn, L.C. 2020 WHO Classification of Female Genital Tumors. Geburtshilfe Frauenheilkd 2021, 81, 1145–1153. [Google Scholar] [CrossRef]
- Danaher, P.; Kim, Y.; Nelson, B.; Griswold, M.; Yang, Z.; Piazza, E.; Beechem, J.M. Advances in mixed cell deconvolution enable quantification of cell types in spatial transcriptomic data. Nat. Commun. 2022, 13, 385. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Herrington, C.S. (Ed.) WHO Classification of Tumours Female Genital Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Griswold, M.; Danaher, P. SpatialDecon: Deconvolution of Mixed Cells from Spatial and/or Bulk Gene Expression Data. R package, Version 1.6.0. 2022. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Agrawal, A.; Balci, H.; Hanspers, K.; Coort, S.L.; Martens, M.; Slenter, D.N.; Ehrhart, F.; Digles, D.; Waagmeester, A.; Wassink, I.; et al. WikiPathways 2024: Next generation pathway database. Nucleic Acids Res. 2024, 52, D679–D689. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef] [PubMed]
- Stur, E.; Corvigno, S.; Xu, M.; Chen, K.; Tan, Y.; Lee, S.; Liu, J.; Ricco, E.; Kraushaar, D.; Castro, P.; et al. Spatially resolved transcriptomics of high-grade serous ovarian carcinoma. iScience 2022, 25, 103923. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Imanaka, S.; Matsubara, S.; Shigetomi, H.; Yoshimoto, C. Dual Role of Tissue Factor Pathway Inhibitor 2—A Novel Serodiagnostic Marker for Ovarian Cancer—In Human Cancers. Int. J. Transl. Med. 2024, 4, 419–438. [Google Scholar] [CrossRef]
- Berntsson, J.; Lundgren, S.; Nodin, B.; Uhlen, M.; Gaber, A.; Jirstrom, K. Expression and prognostic significance of the polymeric immunoglobulin receptor in epithelial ovarian cancer. J. Ovarian Res. 2014, 7, 26. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, N.; Huang, J.; Buckanovich, R.J.; Liang, S.; Barchetti, A.; Vezzani, C.; O’Brien-Jenkins, A.; Wang, J.; Ward, M.R.; et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005, 65, 925–932. [Google Scholar] [CrossRef]
- Wen, T.; Barham, W.; Li, Y.; Zhang, H.; Gicobi, J.K.; Hirdler, J.B.; Liu, X.; Ham, H.; Peterson Martinez, K.E.; Lucien, F.; et al. NKG7 Is a T-cell-Intrinsic Therapeutic Target for Improving Antitumor Cytotoxicity and Cancer Immunotherapy. Cancer Immunol. Res. 2022, 10, 162–181. [Google Scholar] [CrossRef]
- Noyes, D.; Bag, A.; Oseni, S.; Semidey-Hurtado, J.; Cen, L.; Sarnaik, A.A.; Sondak, V.K.; Adeegbe, D. Tumor-associated Tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2022, 10, e004605. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Baras, A.S.; Drake, C.; Liu, J.J.; Gandhi, N.; Kates, M.; Hoque, M.O.; Meeker, A.; Hahn, N.; Taube, J.M.; Schoenberg, M.P.; et al. The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 2016, 5, e1134412. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ueyama, A.; Funaki, S.; Jinushi, K.; Higuchi, N.; Morihara, H.; Hirata, M.; Nagira, Y.; Saito, T.; Kawashima, A.; et al. In situ analysis of CCR8+ regulatory T cells in lung cancer: Suppression of GzmB+ CD8+ T cells and prognostic marker implications. BMC Cancer 2024, 24, 627. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.; Rayman, P.; Uzzo, R.; Clark, P.; Kim, H.J.; Tubbs, R.; Novick, A.; Bukowski, R.; Hamilton, T.; Finke, J. Impaired activation of NFkappaB in T cells from a subset of renal cell carcinoma patients is mediated by inhibition of phosphorylation and degradation of the inhibitor, IkappaBalpha. Blood 1998, 92, 1334–1341. [Google Scholar] [CrossRef]
- Hovelmeyer, N.; Schmidt-Supprian, M.; Ohnmacht, C. NF-kappaB in control of regulatory T cell development, identity, and function. J. Mol. Med. 2022, 100, 985–995. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age Group at Dx | Primary Site | Stage | Tumor Size | PFS > 5y | Recurrence Status | Lymph Node Metastasis | Distant Metastasis |
|---|---|---|---|---|---|---|---|---|
| Pat05 | 70s | Ovary | 3c | 14 | Yes | Non-recurred | Absent | Absent |
| Pat09 | 40s | Ovary | 3c | 5.3 | No | Recurred | Absent | Absent |
| Pat10 | 50s | Ovary | 3c | 7 | No | Recurred | Present | Absent |
| Pat11 | 60s | Ovary | 3b | 6 | Yes | Non-recurred | Absent | Absent |
| Pat12 | 50s | Ovary | 2c | 7.5 | Yes | Non-recurred | Absent | Absent |
| Pat13 | 30s | Ovary | 3c | 4 | Yes | Non-recurred | Present | Absent |
| Pat15 | 50s | Ovary | 3c | 10 | No | Recurred | Present | Absent |
| Pat16 | 50s | Ovary | 3c | 12 | No | Recurred | Present | Absent |
| Pat20 | 50s | Ovary | 3c | 10 | No | Recurred | Present | Absent |
| Pat21 | 50s | Ovary | 3c | 12 | No | Recurred | Present | Absent |
| Pat42 | 50s | Ovary | 3c | 15 | No | Recurred | Present | Absent |
| Pat53 | 60s | Ovary | 3c | 12 | No | Recurred | Present | Absent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Joung, J.-G.; Park, H.; Choi, M.C.; Koh, D.; Jeong, J.-Y.; Lee, J.; Kim, S.-Y.; Jung, D.; Hwang, S.; et al. Identification of Prognostic Biomarkers of Ovarian High-Grade Serous Carcinoma: A Preliminary Study Using Spatial Transcriptome Analysis and Multispectral Imaging. Cells 2025, 14, 681. https://doi.org/10.3390/cells14100681
Kang H, Joung J-G, Park H, Choi MC, Koh D, Jeong J-Y, Lee J, Kim S-Y, Jung D, Hwang S, et al. Identification of Prognostic Biomarkers of Ovarian High-Grade Serous Carcinoma: A Preliminary Study Using Spatial Transcriptome Analysis and Multispectral Imaging. Cells. 2025; 14(10):681. https://doi.org/10.3390/cells14100681
Chicago/Turabian StyleKang, Haeyoun, Je-Gun Joung, Hyun Park, Min Chul Choi, Doohyun Koh, Ju-Yeon Jeong, Jimin Lee, Sook-Young Kim, Daun Jung, Sohyun Hwang, and et al. 2025. "Identification of Prognostic Biomarkers of Ovarian High-Grade Serous Carcinoma: A Preliminary Study Using Spatial Transcriptome Analysis and Multispectral Imaging" Cells 14, no. 10: 681. https://doi.org/10.3390/cells14100681
APA StyleKang, H., Joung, J.-G., Park, H., Choi, M. C., Koh, D., Jeong, J.-Y., Lee, J., Kim, S.-Y., Jung, D., Hwang, S., & An, H. J. (2025). Identification of Prognostic Biomarkers of Ovarian High-Grade Serous Carcinoma: A Preliminary Study Using Spatial Transcriptome Analysis and Multispectral Imaging. Cells, 14(10), 681. https://doi.org/10.3390/cells14100681