
Functional Selectivity of Cannabinoid Type 1 G Protein-Coupled Receptor Agonists in Transactivating Glycosylated Receptors on Cancer Cells to Induce Epithelial–Mesenchymal Transition Metastatic Phenotype

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents
2.3. CB1 Agonist Treatment Protocol (Time and Dosage)
2.4. Antibodies
2.5. Sialidase Assay
2.6. NF-kB Dependent Secreted Embryonic Alkaline Phosphatase (SEAP) Assay
2.7. Co-Localization
2.8. Immunofluorescence Staining
2.9. Statistics
3. Results
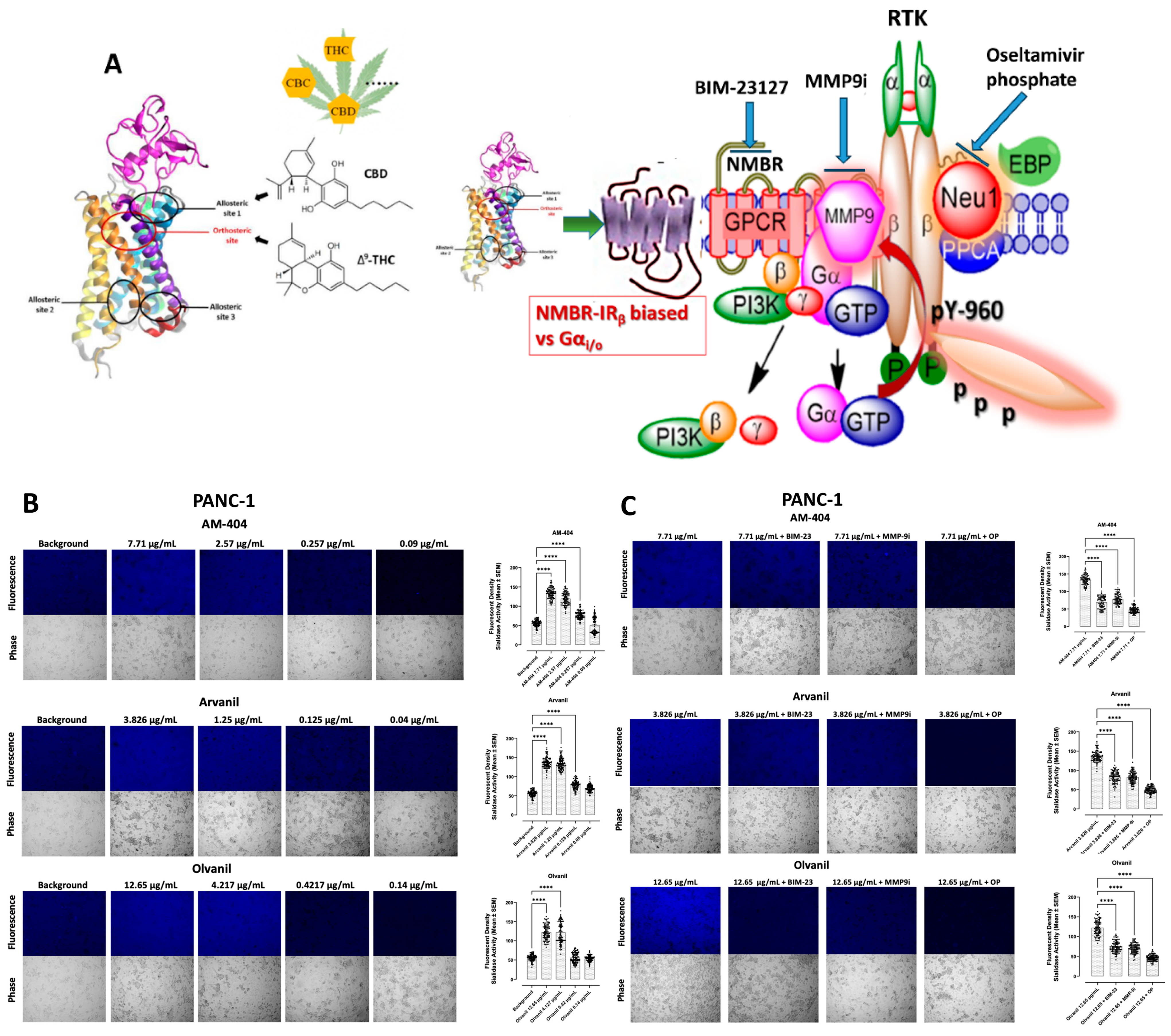
3.1. CB1 G Protein-Coupled Receptor Agonists Dose-Dependently Induce Neu1 Sialidase Activity in RAW-Blue Macrophage Cells

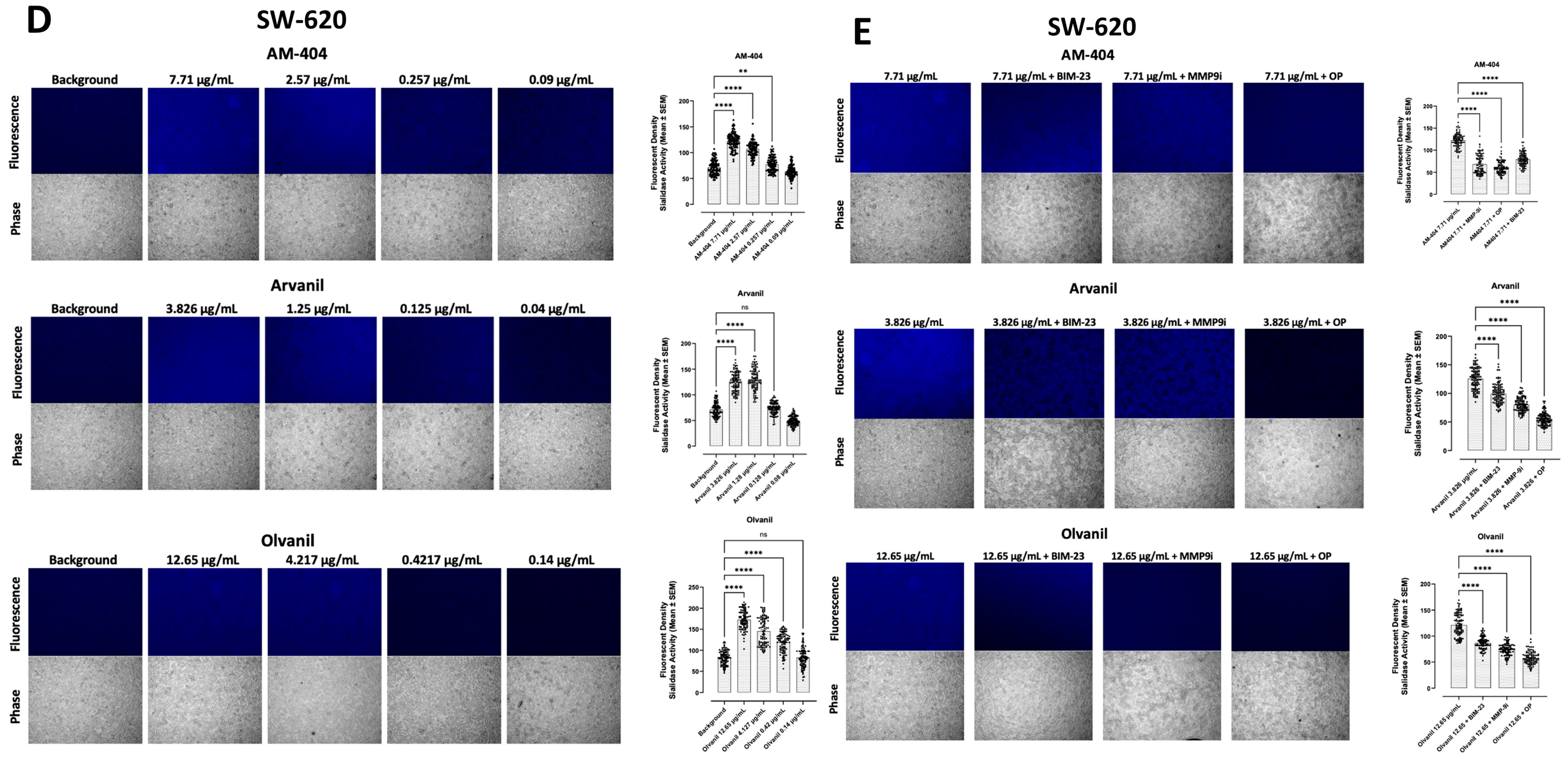
3.2. CB1 G Protein-Coupled Receptor Agonists Dose-Dependently Induce Neu1 Sialidase Activity in Pancreatic PANC-1 and Colorectal SW-620 Cancer Cell Lines
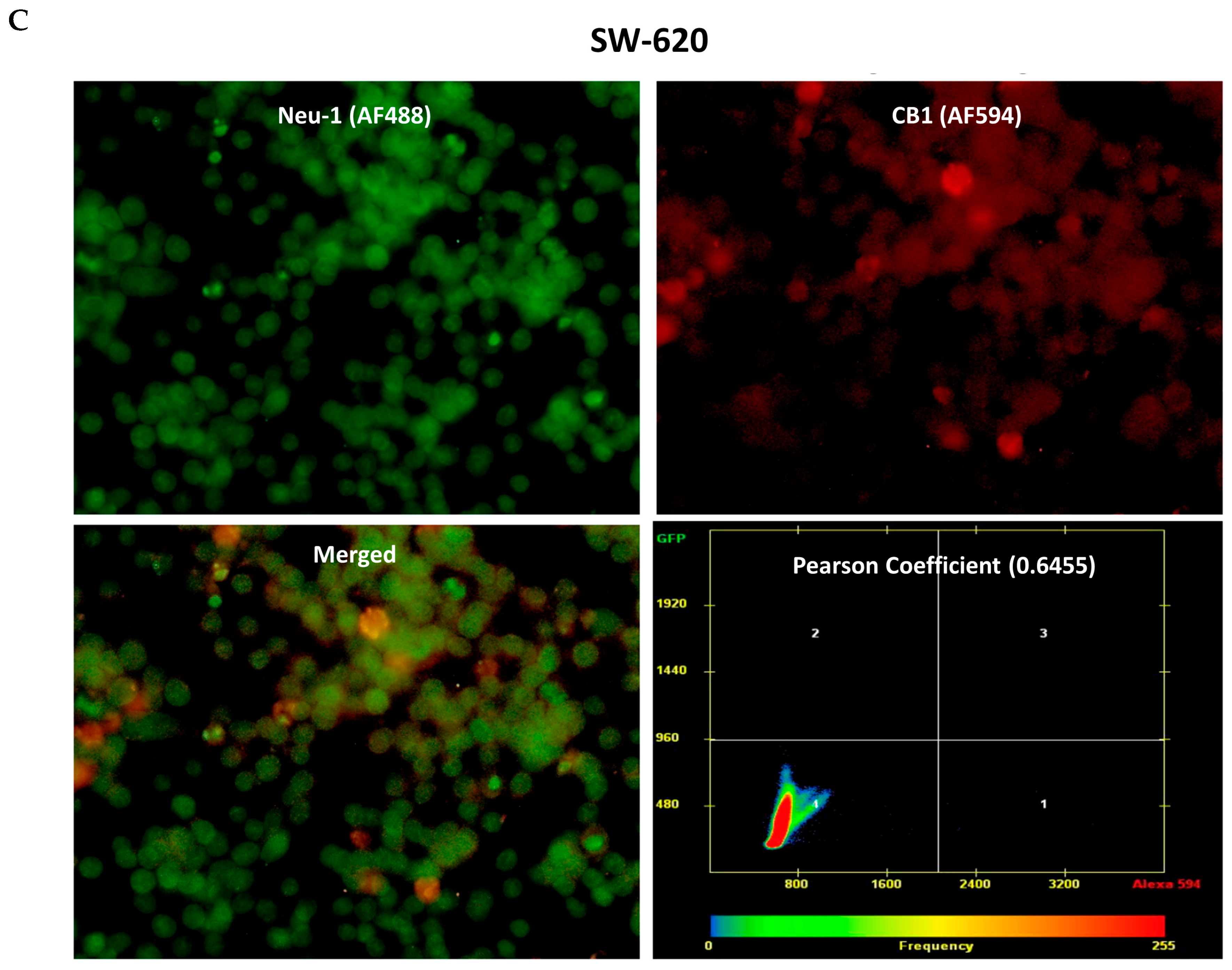
3.3. CB1 Receptor Co-Localizes with Neu1 on the Cell Surface of Naïve Unstimulated RAW-Blue Macrophages, PANC-1, and SW-620 Cells
3.4. Synthetic CB1 Cannabinoids AM-404, Aravnil, and Olvanil Marginally Reduce the Expression of E-Cadherin in SW-620 Colorectal Cancer Cells
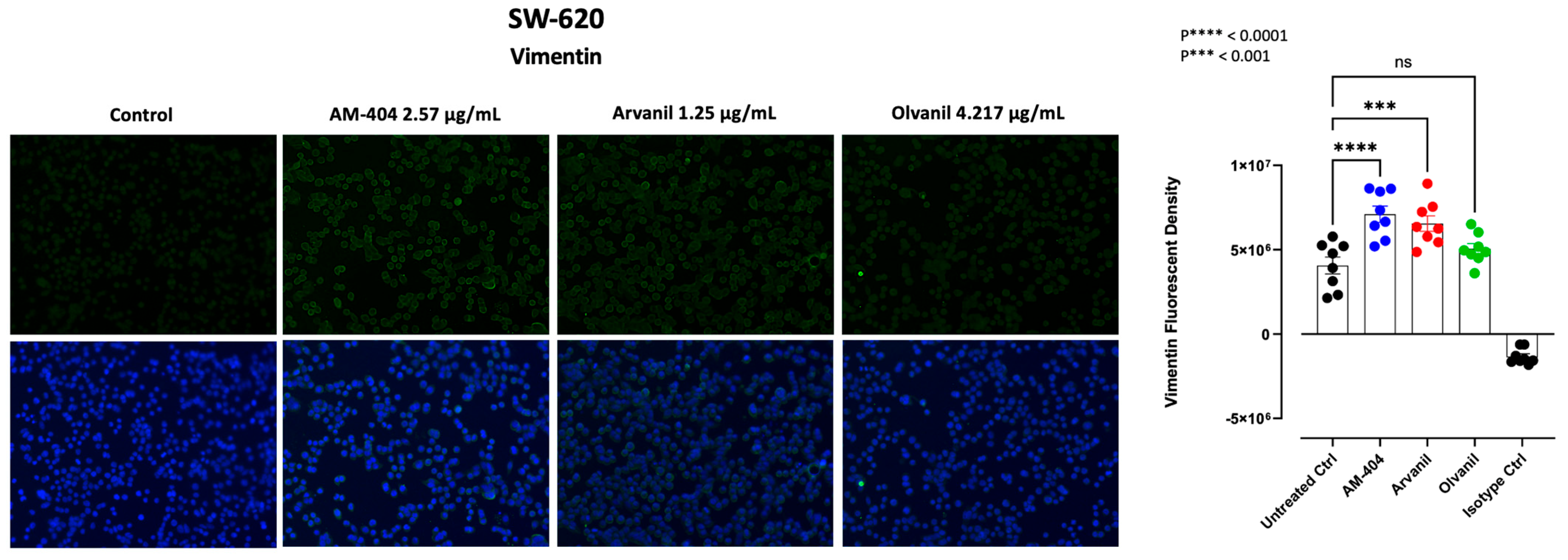
3.5. Synthetic CB1 Cannabinoids AM-404, Arvanil, and Olvanil Significantly Upregulate the Expression of Vimentin in SW-620 Cells
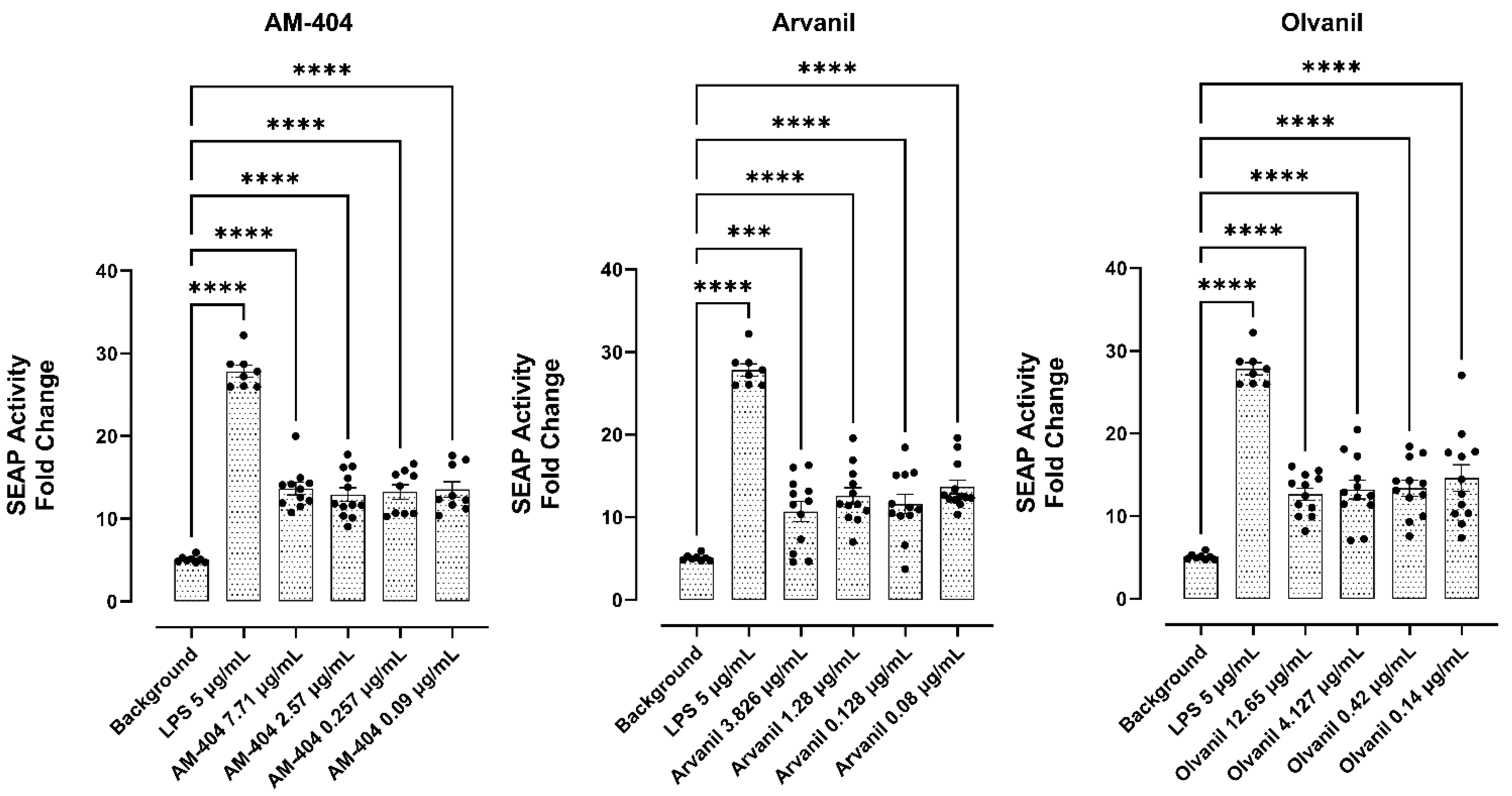
3.6. CB1 Agonists, AM-404, Arvanil, and Olvanil Induce Upregulation of NF-kB
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.H.; Catt, K.J. GPCR-mediated transactivation of RTKs in the CNS: Mechanisms and consequences. Trends Neurosci. 2004, 27, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Delcourt, N.; Bockaert, J.; Marin, P. GPCR-jacking: From a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci. 2007, 28, 602–607. [Google Scholar] [CrossRef]
- Jayanth, P.; Amith, S.R.; Gee, K.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell. Signal. 2010, 22, 1193–1205. [Google Scholar] [CrossRef]
- Alghamdi, F.; Guo, M.; Abdulkhalek, S.; Crawford, N.; Amith, S.R.; Szewczuk, M.R. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell. Signal. 2014, 26, 1355–1368. [Google Scholar] [CrossRef]
- Haxho, F.; Haq, S.; Szewczuk, M.R. Biased G protein-coupled receptor agonism mediates Neu1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell. Signal. 2018, 43, 71–84. [Google Scholar] [CrossRef]
- Haxho, F.; Neufeld, R.J.; Szewczuk, M.R. Neuraminidase-1: A novel therapeutic target in multistage tumorigenesis. Oncotarget 2016, 7, 40860–40881. [Google Scholar] [CrossRef]
- Haxho, F.; Allison, S.; Alghamdi, F.; Brodhagen, L.; Kuta, V.E.L.; Abdulkhalek, S.; Neufeld, R.J.; Szewczuk, M.R. Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma. Breast Cancer Targets Ther. 2014, 6, 191. [Google Scholar]
- Gilmour, A.M.; Abdulkhalek, S.; Cheng, T.S.; Alghamdi, F.; Jayanth, P.; O’Shea, L.K.; Geen, O.; Arvizu, L.A.; Szewczuk, M.R. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell. Signal. 2013, 25, 2587–2603. [Google Scholar] [CrossRef]
- Abdulkhalek, S.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk regulates nucleic acid-induced endosomal TOLL-like receptor-7 and-9 activation, cellular signaling and pro-inflammatory responses. Cell. Signal. 2013, 25, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Amith, S.R.; Franchuk, S.L.; Jayanth, P.; Guo, M.; Finlay, T.; Gilmour, A.; Guzzo, C.; Gee, K.; Beyaert, R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling. J. Biol. Chem. 2011, 286, 36532–36549. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Guo, M.; Amith, S.R.; Jayanth, P.; Szewczuk, M.R. G-protein coupled receptor agonists mediate Neu1 sialidase and matrix metalloproteinase-9 cross-talk to induce transactivation of TOLL-like receptors and cellular signaling. Cell. Signal. 2012, 24, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R.; Gupta, A.; Gagnidze, K.; Lim, M.P.; Gomes, I.; Lee-Ramos, D.; Nieto, N.; Devi, L.A. AT1R-CB₁R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. Embo J. 2011, 30, 2350–2363. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Benito, S.; Moreno, E.; Seijo-Vila, M.; Tundidor, I.; Andradas, C.; Caffarel, M.M.; Caro-Villalobos, M.; Urigüen, L.; Diez-Alarcia, R.; Moreno-Bueno, G.; et al. Therapeutic targeting of HER2-CB(2)R heteromers in HER2-positive breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Wager-Miller, J.; Westenbroek, R.; Mackie, K. Dimerization of G protein-coupled receptors: CB1 cannabinoid receptors as an example. Chem. Phys. Lipids 2002, 121, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kearn, C.; Mackie, K.; Glass, M. Physical interactions of CB1 cannabinoid and D2 receptors. In Proceedings of the 2004 Symposium of the Cannabinoids, Paestum, Italy, 22–27 June 2004; p. 16. [Google Scholar]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef]
- Jarrahian, A.; Watts, V.J.; Barker, E.L. D2 dopamine receptors modulate Gα-subunit coupling of the CB1 cannabinoid receptor. J. Pharmacol. Exp. Ther. 2004, 308, 880–886. [Google Scholar] [CrossRef]
- Hojo, M.; Sudo, Y.; Ando, Y.; Minami, K.; Takada, M.; Matsubara, T.; Kanaide, M.; Taniyama, K.; Sumikawa, K.; Uezono, Y. μ-Opioid receptor forms a functional heterodimer with cannabinoid CB1 receptor: Electrophysiological and FRET assay analysis. J. Pharmacol. Sci. 2008, 108, 308–319. [Google Scholar] [CrossRef]
- Carriba, P.; Ortiz, O.; Patkar, K.; Justinova, Z.; Stroik, J.; Themann, A.; Müller, C.; Woods, A.S.; Hope, B.T.; Ciruela, F. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology 2007, 32, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.D.; Hébert, T.E.; Kelly, M.E. Physical and functional interaction between CB1 cannabinoid receptors and beta2-adrenoceptors. Br. J. Pharmacol. 2010, 160, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Northup, J.K. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1999, 56, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Villaseca, S.; Romero, G.; Ruiz, M.J.; Pérez, C.; Leal, J.I.; Tovar, L.M.; Torrejón, M. Gαi protein subunit: A step toward understanding its non-canonical mechanisms. Front. Cell Dev. Biol. 2022, 10, 941870. [Google Scholar] [CrossRef]
- Hudson, B.D.; Hebert, T.E.; Kelly, M.E. Ligand- and heterodimer-directed signaling of the CB(1) cannabinoid receptor. Mol. Pharmacol. 2010, 77, 1–9. [Google Scholar] [CrossRef]
- Carriba, P.; Navarro, G.; Ciruela, F.; Ferré, S.; Casadó, V.; Agnati, L.; Cortés, A.; Mallol, J.; Fuxe, K.; Canela, E.I. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 2008, 5, 727–733. [Google Scholar] [CrossRef]
- Peeri, H.; Koltai, H. Cannabis Biomolecule Effects on Cancer Cells and Cancer Stem Cells: Cytotoxic, Anti-Proliferative, and Anti-Migratory Activities. Biomolecules 2022, 12, 491. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Preet, A.; Ganju, R.K.; Groopman, J.E. Delta9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene 2008, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Salazar, M.; Carracedo, A.; Lorente, M.; Egia, A.; González-Feria, L.; Haro, A.; Velasco, G.; Guzmán, M. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008, 68, 1945–1952. [Google Scholar] [CrossRef]
- Moon, K.-Y.; Hahn, B.-S.; Lee, J.; Kim, Y.S. A Cell-Based Assay System for Monitoring NF-κB Activity in Human HaCaT Transfectant Cells. Anal. Biochem. 2001, 292, 17–21. [Google Scholar] [CrossRef]
- Hillard, C.J.; Huang, H.; Vogt, C.D.; Rodrigues, B.E.; Neumann, T.S.; Sem, D.S.; Schroeder, F.; Cunningham, C.W. Endocannabinoid Transport Proteins: Discovery of Tools to Study Sterol Carrier Protein-2. Methods Enzym. 2017, 593, 99–121. [Google Scholar] [CrossRef]
- Marzęda, P.; Wróblewska-Łuczka, P.; Florek-Łuszczki, M.; Drozd, M.; Góralczyk, A.; Łuszczki, J.J. Comparison of the Anticancer Effects of Arvanil and Olvanil When Combined with Cisplatin and Mitoxantrone in Various Melanoma Cell Lines-An Isobolographic Analysis. Int. J. Mol. Sci. 2022, 23, 4192. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Bisogno, T.; Davis, J.B.; Pertwee, R.G.; Di Marzo, V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: Inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000, 483, 52–56. [Google Scholar] [CrossRef]
- Fegley, D.; Kathuria, S.; Mercier, R.; Li, C.; Goutopoulos, A.; Makriyannis, A.; Piomelli, D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc. Natl. Acad. Sci. USA 2004, 101, 8756–8761. [Google Scholar] [CrossRef] [PubMed]
- Ralevic, V.; Kendall, D.A.; Jerman, J.C.; Middlemiss, D.N.; Smart, D. Cannabinoid activation of recombinant and endogenous vanilloid receptors. Eur. J. Pharmacol. 2001, 424, 211–219. [Google Scholar] [CrossRef]
- López-Rodríguez, M.L.; Viso, A.; Ortega-Gutiérrez, S.; Lastres-Becker, I.; González, S.; Fernández-Ruiz, J.; Ramos, J.A. Design, synthesis and biological evaluation of novel arachidonic acid derivatives as highly potent and selective endocannabinoid transporter inhibitors. J. Med. Chem. 2001, 44, 4505–4508. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Chuang, H.-h.; Movahed, P.; Julius, D.; Högestätt, E.D. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur. J. Pharmacol. 2000, 396, 39–42. [Google Scholar] [CrossRef]
- Glaser, S.T.; Abumrad, N.A.; Fatade, F.; Kaczocha, M.; Studholme, K.M.; Deutsch, D.G. Evidence against the presence of an anandamide transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 4269–4274. [Google Scholar] [CrossRef]
- Melck, D.; Bisogno, T.; De Petrocellis, L.; Chuang, H.-h.; Julius, D.; Bifulco, M.; Di Marzo, V. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): Vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Commun. 1999, 262, 275–284. [Google Scholar] [CrossRef]
- Sancho, R.; De La Vega, L.; Appendino, G.; Di Marzo, V.; Macho, A.; Muñoz, E. The CB1/VR1 agonist arvanil induces apoptosis through an FADD/caspase-8-dependent pathway. Br. J. Pharmacol. 2003, 140, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Bisogno, T.; Melck, D.; Ross, R.; Brockie, H.; Stevenson, L.; Pertwee, R.; De Petrocellis, L. Interactions between synthetic vanilloids and the endogenous cannabinoid system. FEBS Lett. 1998, 436, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Piomelli, D. Anandamide transport inhibition by the vanilloid agonist olvanil. Eur. J. Pharmacol. 1999, 364, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Leelawat, S.; Leelawat, K.; Narong, S.; Matangkasombut, O. The dual effects of Δ9-tetrahydrocannabinol on cholangiocarcinoma cells: Anti-invasion activity at low concentration and apoptosis induction at high concentration. Cancer Investig. 2010, 28, 357–363. [Google Scholar] [CrossRef]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress–related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef] [PubMed]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Au-Amith, S.R.; Au-Jayanth, P.; Au-Finlay, T.; Au-Franchuk, S.; Au-Gilmour, A.; Au-Abdulkhalek, S.; Au-Szewczuk, M.R. Detection of Neu1 Sialidase Activity in Regulating Toll-like Receptor Activation. JoVE 2010, 43, e2142. [Google Scholar] [CrossRef]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Siddiqui, S.; Seyrantepe, V.; Gee, K.; Basta, S.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Dependence of pathogen molecule-induced Toll-like receptor activation and cell function on Neu1 sialidase. Glycoconj. J. 2009, 26, 1197. [Google Scholar] [CrossRef]
- O’Shea, L.K.; Abdulkhalek, S.; Allison, S.; Neufeld, R.J.; Szewczuk, M.R. Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu®) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance. Onco Targets Ther. 2014, 7, 117–134. [Google Scholar] [CrossRef]
- Bunsick, D.A.; Matsukubo, J.; Szewczuk, M.R. Cannabinoids Transmogrify Cancer Metabolic Phenotype via Epigenetic Reprogramming and a Novel CBD Biased G Protein-Coupled Receptor Signaling Platform. Cancers 2023, 15, 1030. [Google Scholar] [CrossRef]
- Herold, C.L.; Behm, D.J.; Buckley, P.T.; Foley, J.J.; Wixted, W.E.; Sarau, H.M.; Douglas, S.A. The neuromedin B receptor antagonist, BIM-23127, is a potent antagonist at human and rat urotensin-II receptors. Br. J. Pharmacol. 2003, 139, 203–207. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Z.; Capó-Aponte, J.E.; Zhang, F.; Pan, Z.; Reinach, P.S. Epidermal growth factor receptor transactivation by the cannabinoid receptor (CB1) and transient receptor potential vanilloid 1 (TRPV1) induces differential responses in corneal epithelial cells. Exp. Eye Res. 2010, 91, 462–471. [Google Scholar] [CrossRef]
- Melck, D.; De Petrocellis, L.; Orlando, P.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–126. [Google Scholar] [CrossRef]
- Duncan, M.; Galic, M.A.; Wang, A.; Chambers, A.P.; McCafferty, D.M.; McKay, D.M.; Sharkey, K.A.; Pittman, Q.J. Cannabinoid 1 receptors are critical for the innate immune response to TLR4 stimulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R224–R231. [Google Scholar] [CrossRef]
- Khajehali, E.; Malone, D.T.; Glass, M.; Sexton, P.M.; Christopoulos, A.; Leach, K. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol. Pharmacol. 2015, 88, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, R.; Morales, P.; Reggio, P.H. Structural Insights into CB1 Receptor Biased Signaling. Int. J. Mol. Sci. 2019, 20, 1837. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Burandt, E.; Lübbersmeyer, F.; Gorbokon, N.; Büscheck, F.; Luebke, A.M.; Menz, A.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; Höflmayer, D.; et al. E-Cadherin expression in human tumors: A tissue microarray study on 10,851 tumors. Biomark. Res. 2021, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Li, N.; Tu, T.; Tao, Y.; Bi, Y.; Yuan, D.; Zhang, N.; Yang, X.; Kong, D.; You, H.; et al. Hepatitis B virus core protein promotes the expression of neuraminidase 1 to facilitate hepatocarcinogenesis. Lab. Investig. 2020, 100, 1602–1617. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Q.; Liu, K.; Shi, N.; Ma, G.; Wang, P.; Xie, H.M.; Jin, S.J.; Wei, T.T.; Yu, X.Y.; Wang, Y.; et al. Neuraminidase 1 promotes renal fibrosis development in male mice. Nat. Commun. 2023, 14, 1713. [Google Scholar] [CrossRef]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of vimentin in health and disease. Genes. Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]
- Berr, A.L.; Wiese, K.; Dos Santos, G.; Koch, C.M.; Anekalla, K.R.; Kidd, M.; Davis, J.M.; Cheng, Y.; Hu, Y.S.; Ridge, K.M. Vimentin is required for tumor progression and metastasis in a mouse model of non-small cell lung cancer. Oncogene 2023, 42, 2074–2087. [Google Scholar] [CrossRef]
- Hewitt, R.E.; McMarlin, A.; Kleiner, D.; Wersto, R.; Martin, P.; Tsokos, M.; Stamp, G.W.; Stetler-Stevenson, W.G. Validation of a model of colon cancer progression. J. Pathol. 2000, 192, 446–454. [Google Scholar] [CrossRef]
- Toussaint, K.; Appert-Collin, A.; Morjani, H.; Albrecht, C.; Sartelet, H.; Romier-Crouzet, B.; Maurice, P.; Duca, L.; Blaise, S.; Bennasroune, A. Neuraminidase-1: A Sialidase Involved in the Development of Cancers and Metabolic Diseases. Cancers 2022, 14, 4868. [Google Scholar] [CrossRef]
- Rodgers, J.; Sundararaj, K.; Bruner, E.; Wolf, B.; Nowling, T.K. The role of neuraminidase 1 (NEU1) in cytokine release by primary mouse mesangial cells and disease outcomes in murine lupus nephritis. Autoimmunity 2021, 54, 163–175. [Google Scholar] [CrossRef]
- Fougerat, A.; Pan, X.; Smutova, V.; Heveker, N.; Cairo, C.W.; Issad, T.; Larrivée, B.; Medin, J.A.; Pshezhetsky, A.V. Neuraminidase 1 activates insulin receptor and reverses insulin resistance in obese mice. Mol. Metab. 2018, 12, 76–88. [Google Scholar] [CrossRef]
- Mackie, K. Cannabinoid receptor homo-and heterodimerization. Life Sci. 2005, 77, 1667–1673. [Google Scholar] [CrossRef]
- Tóth, A.D.; Turu, G.; Hunyady, L.; Balla, A. Novel mechanisms of G-protein-coupled receptors functions: AT1 angiotensin receptor acts as a signaling hub and focal point of receptor cross-talk. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 69–82. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.-T.; Hang, L.; Liu, T. Mu opioid receptor heterodimers emerge as novel therapeutic targets: Recent progress and future perspective. Front. Pharmacol. 2020, 11, 1078. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Marini, P.; Matias, I.; Moriello, A.S.; Starowicz, K.; Cristino, L.; Nigam, S.; Di Marzo, V. Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma β-cells. Exp. Cell Res. 2007, 313, 2993–3004. [Google Scholar] [CrossRef]
- Ohki-Hamazaki, H.; Wada, E.; Matsui, K.; Wada, K. Cloning and expression of the neuromedin B receptor and the third subtype of bombesin receptor genes in the mouse. Brain Res. 1997, 762, 165–172. [Google Scholar] [CrossRef]
- Ohki-Hamazaki, H.; Sakai, Y.; Kamata, K.; Ogura, H.; Okuyama, S.; Watase, K.; Yamada, K.; Wada, K. Functional properties of two bombesin-like peptide receptors revealed by the analysis of mice lacking neuromedin B receptor. J. Neurosci. 1999, 19, 948–954. [Google Scholar] [CrossRef][Green Version]
- Dalton, G.D.; Howlett, A.C. Cannabinoid CB1 receptors transactivate multiple receptor tyrosine kinases and regulate serine/threonine kinases to activate ERK in neuronal cells. Br. J. Pharmacol. 2012, 165, 2497–2511. [Google Scholar] [CrossRef]
- Leifer, C.A.; Medvedev, A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef]
- Leo, L.M.; Abood, M.E. CB1 Cannabinoid Receptor Signaling and Biased Signaling. Molecules 2021, 26, 5413. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef]
- Luo, C.K.; Chou, P.H.; Ng, S.K.; Lin, W.Y.; Wei, T.T. Cannabinoids orchestrate cross-talk between cancer cells and endothelial cells in colorectal cancer. Cancer Gene Ther. 2022, 29, 597–611. [Google Scholar] [CrossRef]
- Rosenthaler, S.; Pöhn, B.; Kolmanz, C.; Huu, C.N.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef]
- Morales, P.; Goya, P.; Jagerovic, N. Emerging strategies targeting CB2 cannabinoid receptor: Biased agonism and allosterism. Biochem. Pharmacol. 2018, 157, 8–17. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Biased type 1 cannabinoid receptor signaling influences neuronal viability in a cell culture model of Huntington disease. Mol. Pharmacol. 2016, 89, 364–375. [Google Scholar] [CrossRef]
- Qorri, B.; Kalaydina, R.V.; Velickovic, A.; Kaplya, Y.; Decarlo, A.; Szewczuk, M.R. Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling. Cells 2018, 7, 117. [Google Scholar] [CrossRef]
- Jakowiecki, J.; Abel, R.; Orzeł, U.; Pasznik, P.; Preissner, R.; Filipek, S. Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol-A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling. Molecules 2021, 26, 2456. [Google Scholar] [CrossRef]
- Reber, L.; Vermeulen, L.; Haegeman, G.; Frossard, N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS ONE 2009, 4, e4393. [Google Scholar] [CrossRef]
- Verstrepen, L.; Bekaert, T.; Chau, T.L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R signaling to NF-κB: Variations on a common theme. Cell. Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Factor, V.M.; Thorgeirsson, S.S. Epigenetic regulation of cancer stem cells in liver cancer: Current concepts and clinical implications. J. Hepatol. 2010, 53, 568–577. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, D., Jr.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a Promising Anti-Cancer Drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Alaaeldin, R.; Ali, F.E.M.; Bekhit, A.A.; Zhao, Q.L.; Fathy, M. Inhibition of NF-kB/IL-6/JAK2/STAT3 Pathway and Epithelial-Mesenchymal Transition in Breast Cancer Cells by Azilsartan. Molecules 2022, 27, 7825. [Google Scholar] [CrossRef]
- Abdel-Latif, R.T.; Wadie, W.; Abdel-Mottaleb, Y.; Abdallah, D.M.; El-Maraghy, N.N.; El-Abhar, H.S. Reposition of the anti-inflammatory drug diacerein in an in-vivo colorectal cancer model. Saudi Pharm. J. 2022, 30, 72–90. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunsick, D.A.; Matsukubo, J.; Aldbai, R.; Baghaie, L.; Szewczuk, M.R. Functional Selectivity of Cannabinoid Type 1 G Protein-Coupled Receptor Agonists in Transactivating Glycosylated Receptors on Cancer Cells to Induce Epithelial–Mesenchymal Transition Metastatic Phenotype. Cells 2024, 13, 480. https://doi.org/10.3390/cells13060480
Bunsick DA, Matsukubo J, Aldbai R, Baghaie L, Szewczuk MR. Functional Selectivity of Cannabinoid Type 1 G Protein-Coupled Receptor Agonists in Transactivating Glycosylated Receptors on Cancer Cells to Induce Epithelial–Mesenchymal Transition Metastatic Phenotype. Cells. 2024; 13(6):480. https://doi.org/10.3390/cells13060480
Chicago/Turabian StyleBunsick, David A., Jenna Matsukubo, Rashelle Aldbai, Leili Baghaie, and Myron R. Szewczuk. 2024. "Functional Selectivity of Cannabinoid Type 1 G Protein-Coupled Receptor Agonists in Transactivating Glycosylated Receptors on Cancer Cells to Induce Epithelial–Mesenchymal Transition Metastatic Phenotype" Cells 13, no. 6: 480. https://doi.org/10.3390/cells13060480
APA StyleBunsick, D. A., Matsukubo, J., Aldbai, R., Baghaie, L., & Szewczuk, M. R. (2024). Functional Selectivity of Cannabinoid Type 1 G Protein-Coupled Receptor Agonists in Transactivating Glycosylated Receptors on Cancer Cells to Induce Epithelial–Mesenchymal Transition Metastatic Phenotype. Cells, 13(6), 480. https://doi.org/10.3390/cells13060480