
Melatonin Improves Glucose Homeostasis and Insulin Sensitivity by Mitigating Inflammation and Activating AMPK Signaling in a Mouse Model of Sleep Fragmentation




Abstract
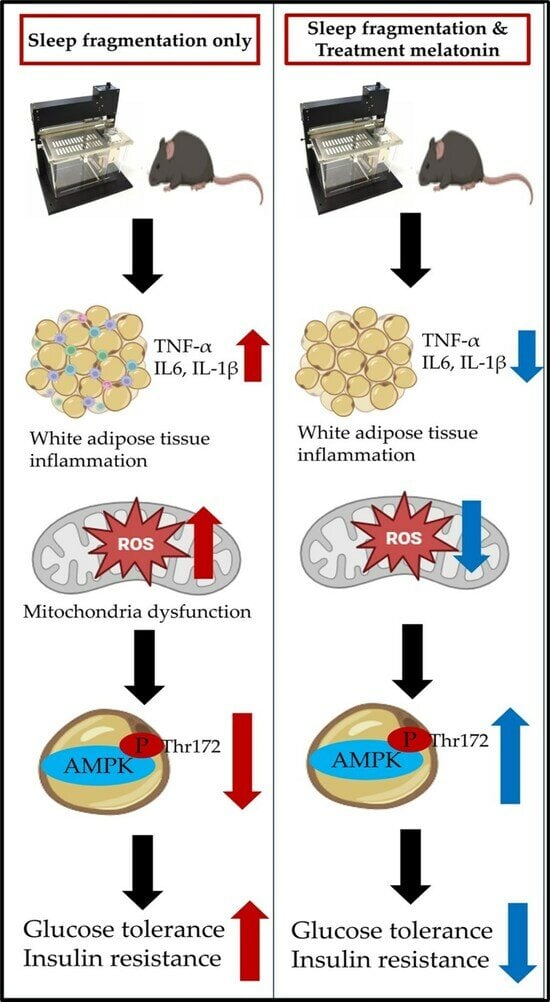
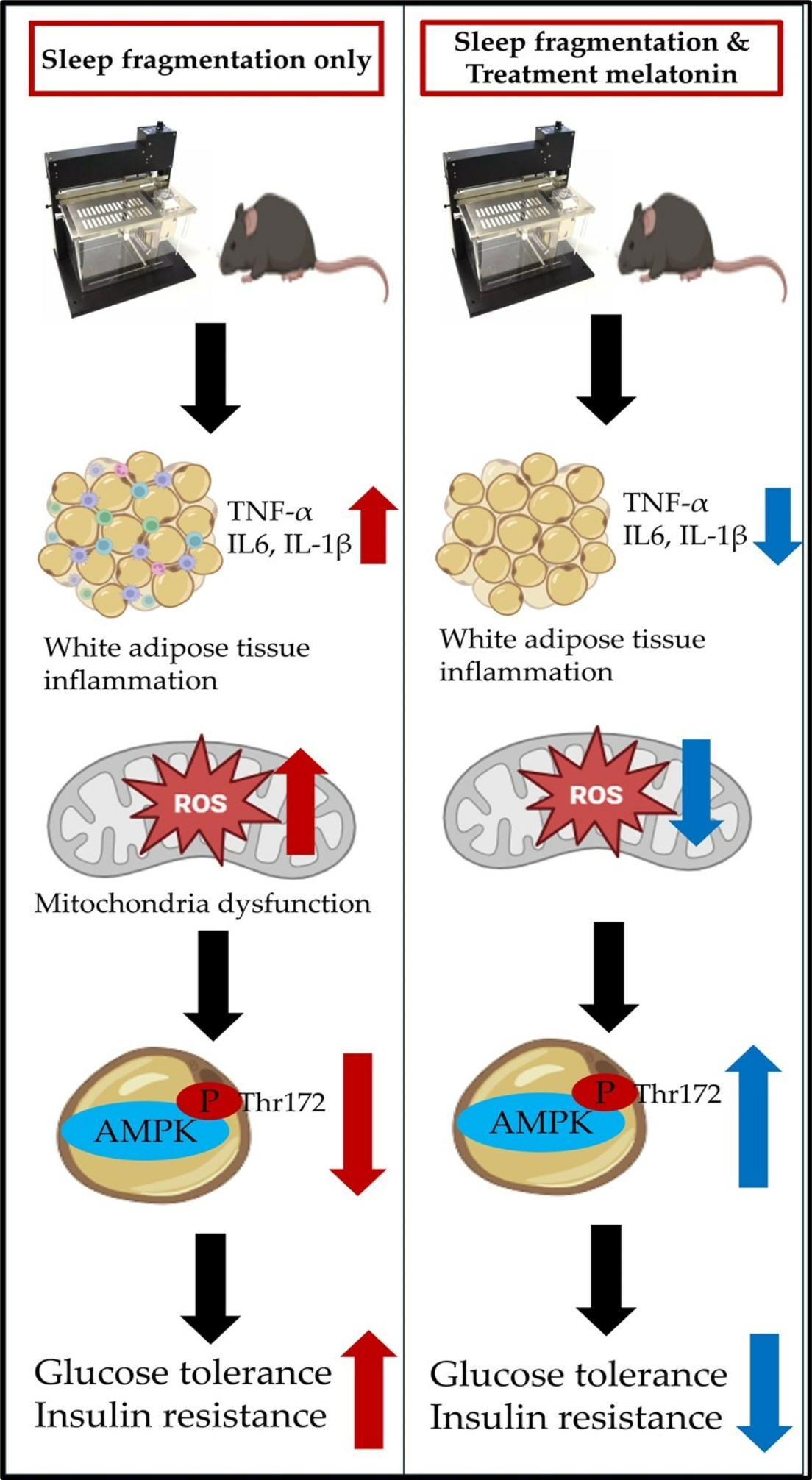{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
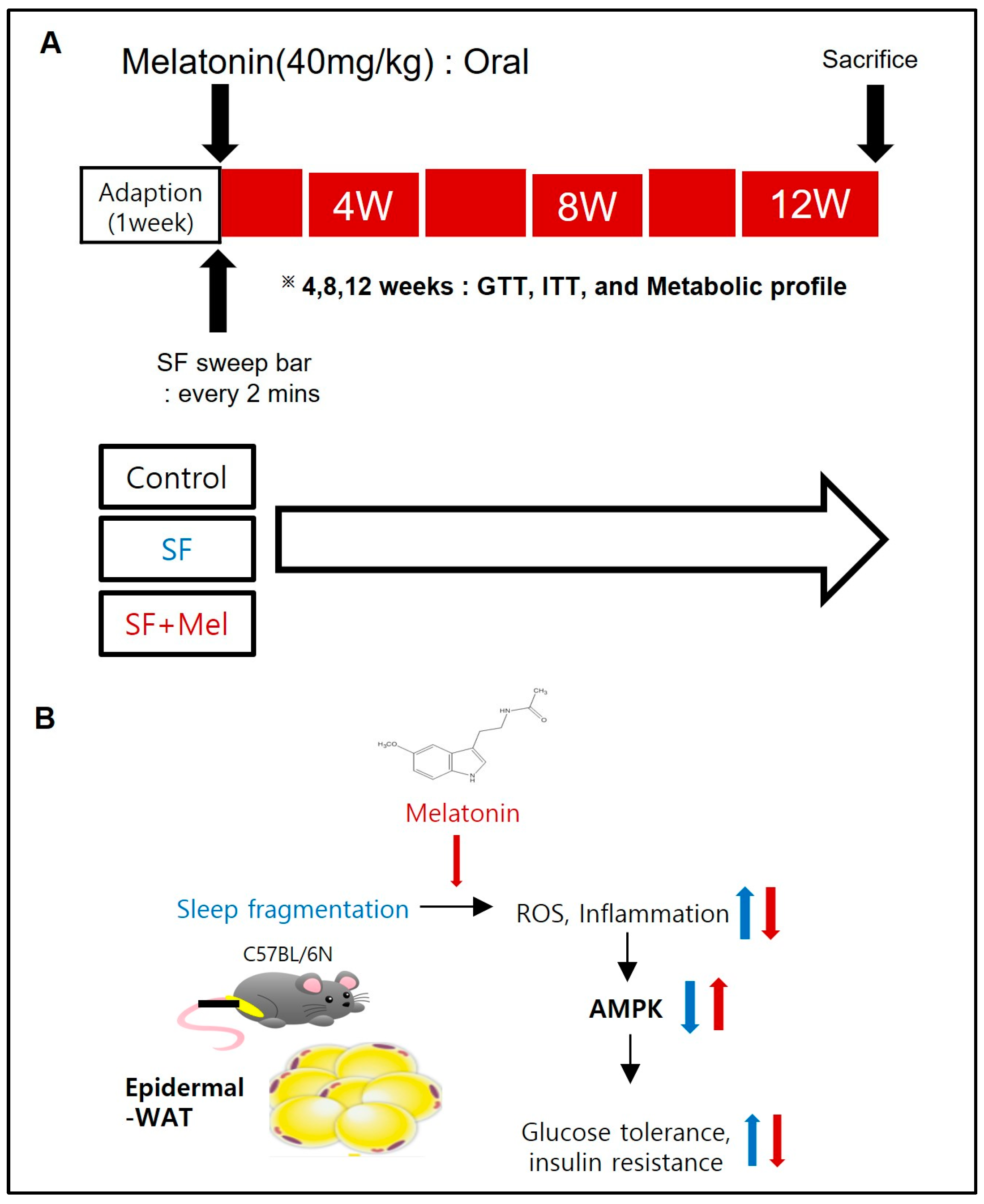
2.1. Schematic Diagram and Graphical Summary of a Mouse Model of SF
2.2. Experiment Design and Animals
2.3. Sleep Fragmentation and Melatonin Treatment
2.4. Biochemical Analysis
2.5. Immunofluorescent Staining for Detecting the AMPK and F4/80 in WAT
2.6. Quantitative Real Time Polymerase Chain Reaction
2.7. Flow Cytometry Analysis and Western Blotting
2.8. Statistical Analysis
3. Results
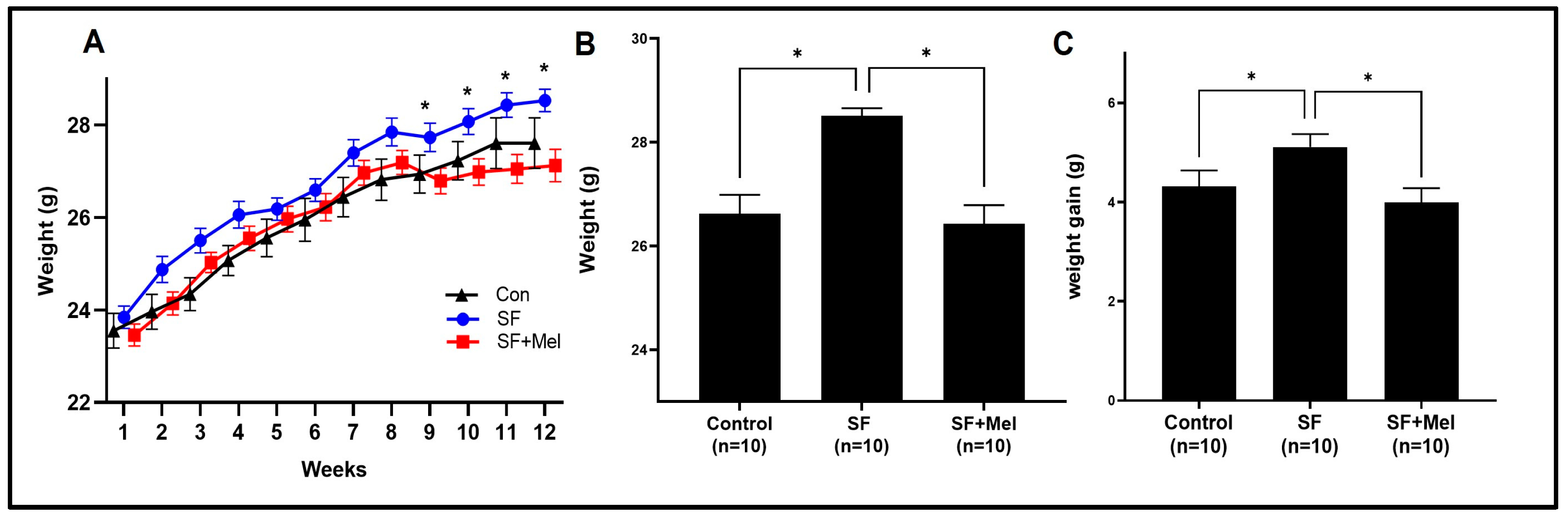
3.1. The Change in Body Weight after Exposure to SF and Treatment with Melatonin in Mice
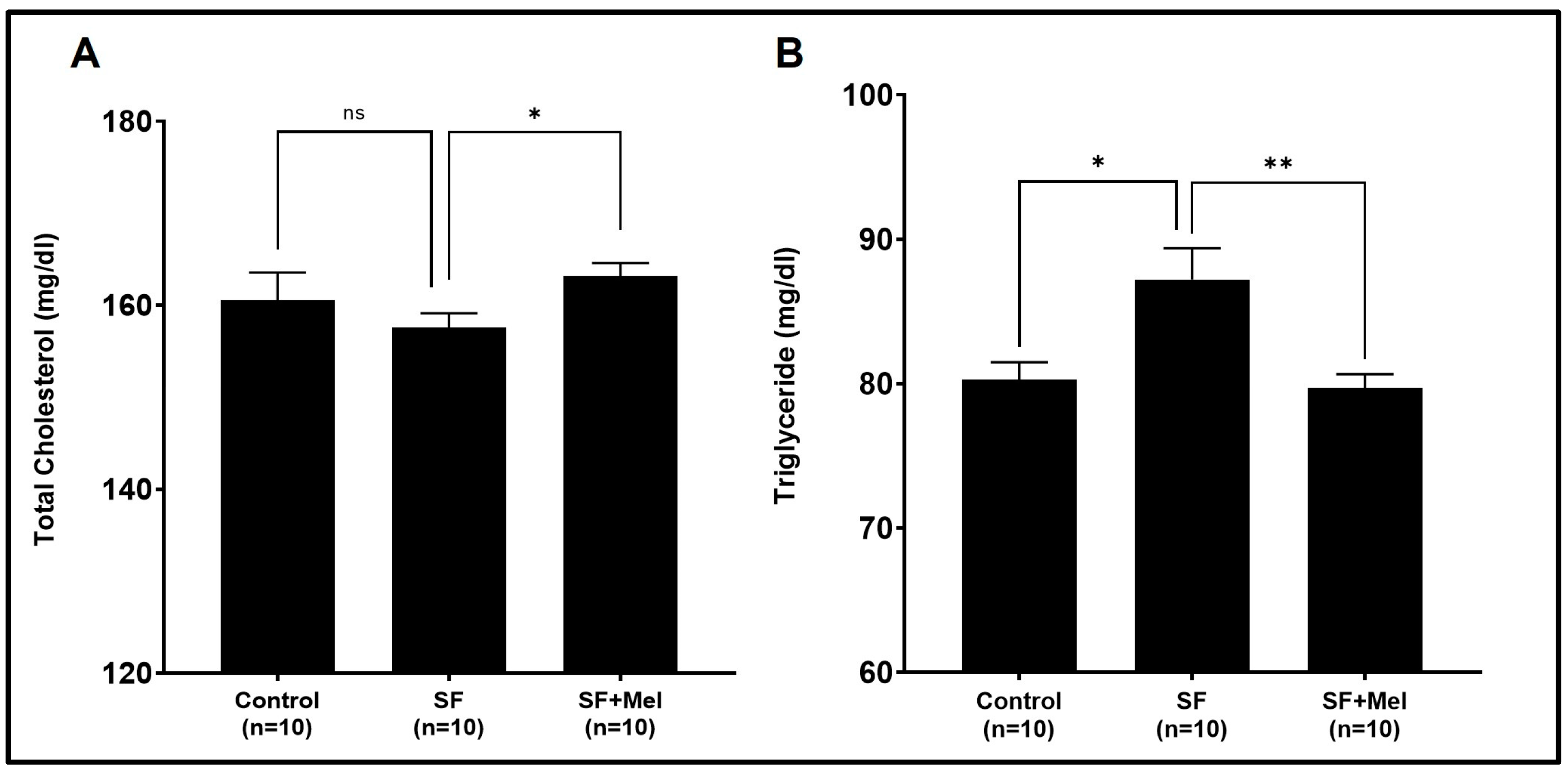
3.2. The Alteration of Lipid Profiles after Exposure to SF and Treatment with Melatonin
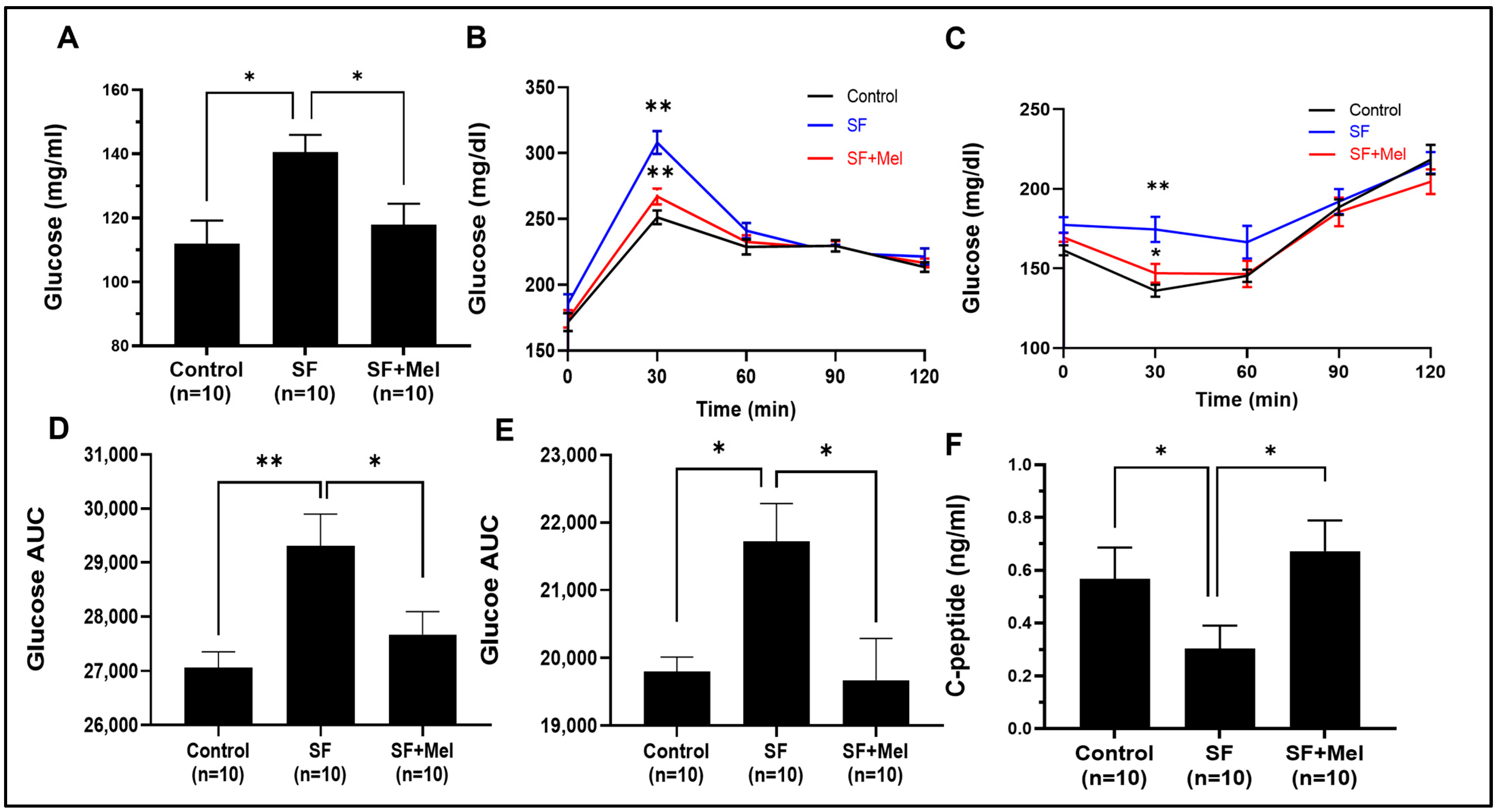
3.3. Melatonin Improved Glycemic Dysregulation in SF Exposed Mice
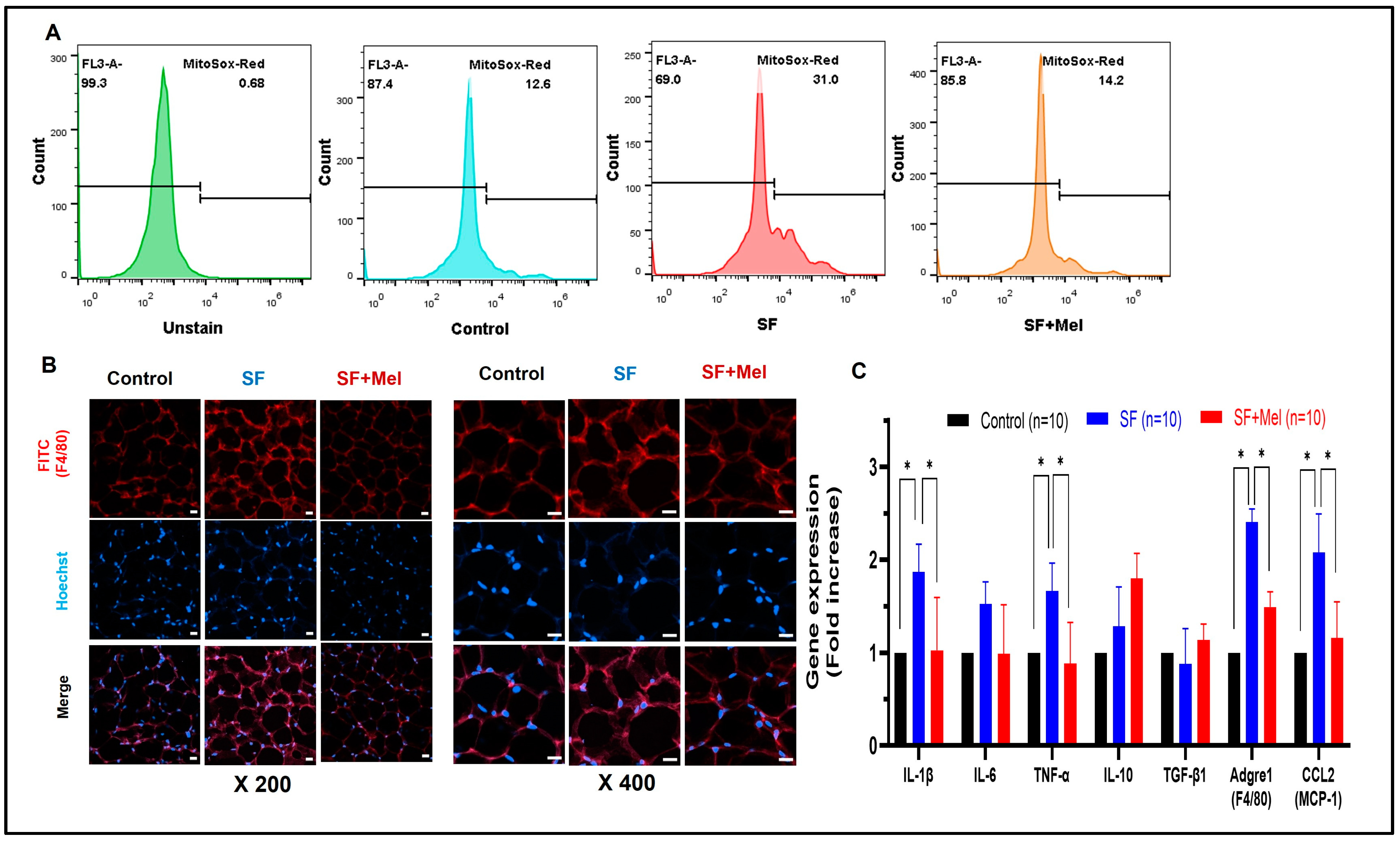
3.4. SF Exhibited Increased ROS and Inflammation in WAT, and These Improved with Melatonin Treatment for 12 Weeks
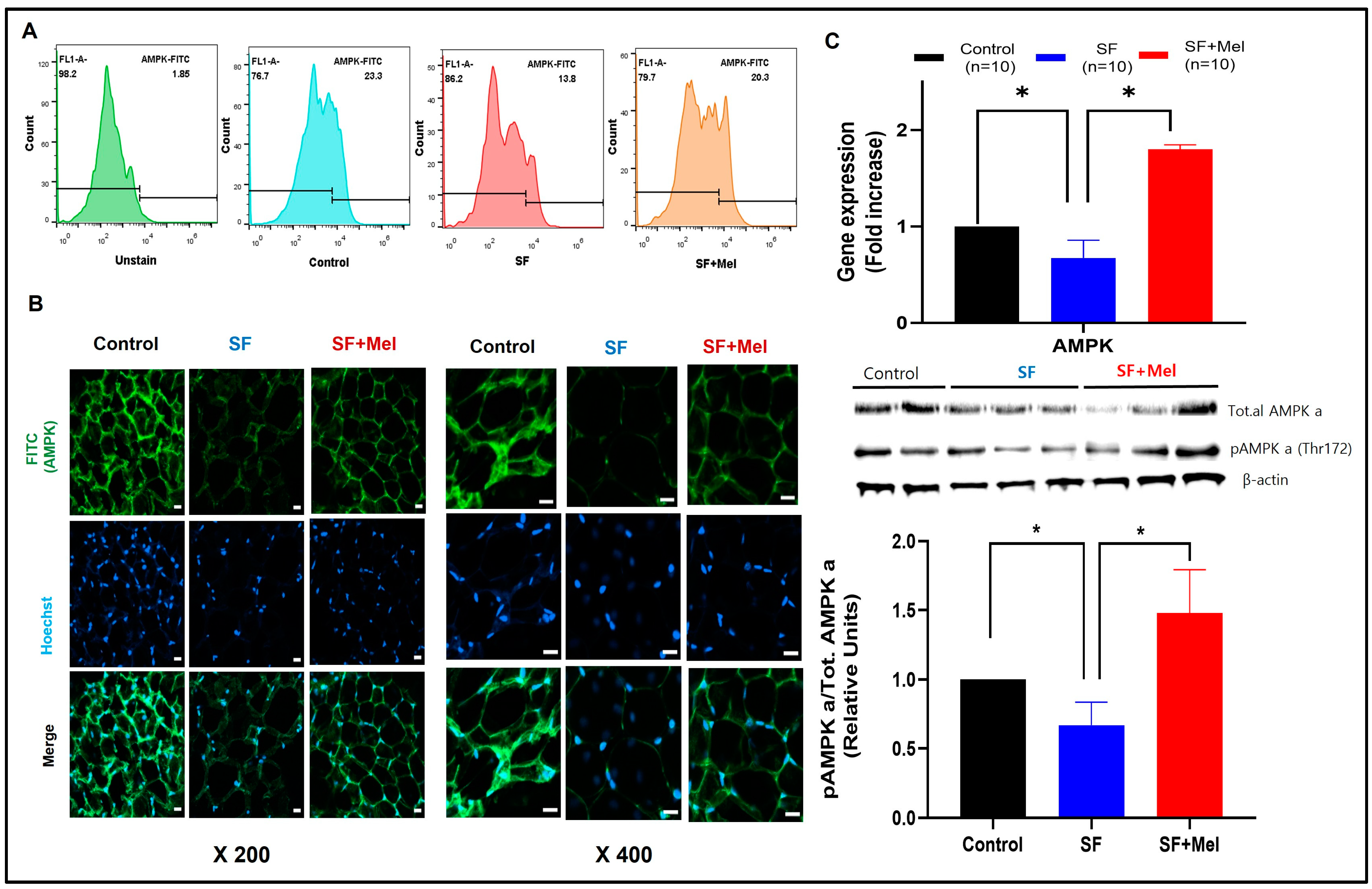
3.5. SF Decreased AMPK Level and Melatonin Treatment Improved AMPK Levels in WAT for 12 Weeks
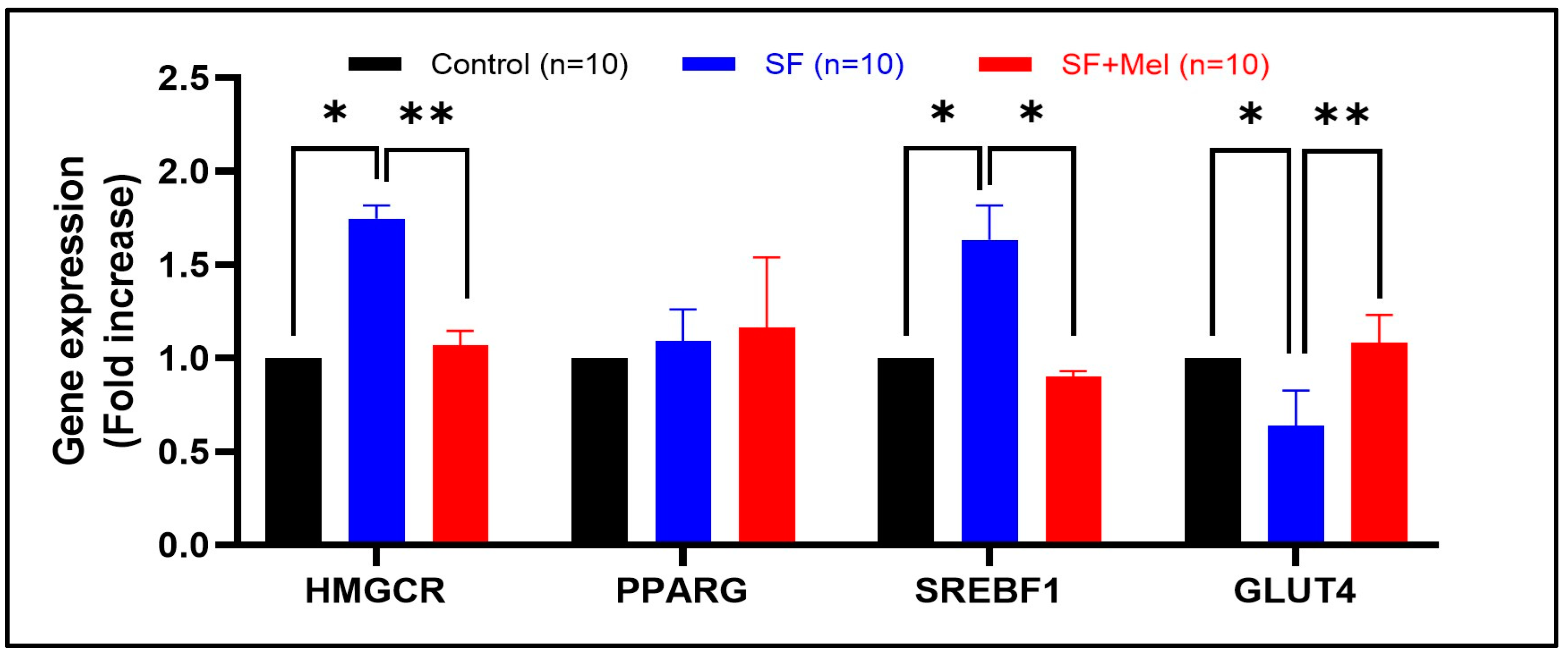
3.6. The Gene Expression Levels of Glycolipid Pathway Associated with AMPK in WAT after Exposure to SF and Melatonin Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, A.M.; Thomas, A.; Crinion, S.J.; Kent, B.D.; Tambuwala, M.M.; Fabre, A.; Pepin, J.L.; Roche, H.M.; Arnaud, C.; Ryan, S. Intermittent hypoxia in obstructive sleep apnoea mediates insulin resistance through adipose tissue inflammation. Eur. Respir. J. 2017, 49, 1601731. [Google Scholar] [CrossRef]
- Aurora, R.N.; Punjabi, N.M. Obstructive sleep apnoea and type 2 diabetes mellitus: A bidirectional association. Lancet Respir. Med. 2013, 1, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Arnaud, C.; Fitzpatrick, S.F.; Gaucher, J.; Tamisier, R.; Pepin, J.L. Adipose tissue as a key player in obstructive sleep apnoea. Eur. Respir. Rev. 2019, 28, 190006. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Gileles-Hillel, A.; Kheirandish-Gozal, L.; Gozal, D. Biological plausibility linking sleep apnoea and metabolic dysfunction. Nat. Rev. Endocrinol. 2016, 12, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Cano Barquilla, P.; Pagano, E.S.; Jimenez-Ortega, V.; Fernandez-Mateos, P.; Esquifino, A.I.; Cardinali, D.P. Melatonin normalizes clinical and biochemical parameters of mild inflammation in diet-induced metabolic syndrome in rats. J. Pineal Res. 2014, 57, 280–290. [Google Scholar] [CrossRef]
- Xu, P.; Wang, J.; Hong, F.; Wang, S.; Jin, X.; Xue, T.; Jia, L.; Zhai, Y. Melatonin prevents obesity through modulation of gut microbiota in mice. J. Pineal Res. 2017, 62, e12399. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. Mechanisms of Melatonin in Obesity: A Review. Int. J. Mol. Sci. 2021, 23, 218. [Google Scholar] [CrossRef]
- Karamitri, A.; Jockers, R. Melatonin in type 2 diabetes mellitus and obesity. Nat. Rev. Endocrinol. 2019, 15, 105–125. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Daval, M.; Foufelle, F.; Ferre, P. Functions of AMP-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lamia, K.A.; Sachdeva, U.M.; DiTacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440. [Google Scholar] [CrossRef]
- Cedernaes, J.; Osler, M.E.; Voisin, S.; Broman, J.E.; Vogel, H.; Dickson, S.L.; Zierath, J.R.; Schioth, H.B.; Benedict, C. Acute Sleep Loss Induces Tissue-Specific Epigenetic and Transcriptional Alterations to Circadian Clock Genes in Men. J. Clin. Endocrinol. Metab. 2015, 100, E1255–E1261. [Google Scholar] [CrossRef] [PubMed]
- Canbolat, E.; Cakiroglu, F.P. The importance of AMPK in obesity and chronic diseases and the relationship of AMPK with nutrition: A literature review. Crit. Rev. Food Sci. Nutr. 2023, 63, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1alpha activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic. Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, D.; Li, H.; Zhang, O.; Huang, Y.; Shao, H.; Wang, Y.; Cai, S.; Zhu, Y.; Jin, S.; et al. Suppression of obesity by melatonin through increasing energy expenditure and accelerating lipolysis in mice fed a high-fat diet. Nutr. Diabetes 2022, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Khalyfa, A.; Wang, Y.; Carreras, A.; Hakim, F.; Neel, B.A.; Brady, M.J.; Qiao, Z.; Hirotsu, C.; Gozal, D. Sleep fragmentation promotes NADPH oxidase 2-mediated adipose tissue inflammation leading to insulin resistance in mice. Int. J. Obes. 2014, 38, 619–624. [Google Scholar] [CrossRef]
- Lee, D.B.; An, S.Y.; Pyo, S.S.; Kim, J.; Kim, S.W.; Yoon, D.W. Sleep Fragmentation Accelerates Carcinogenesis in a Chemical-Induced Colon Cancer Model. Int. J. Mol. Sci. 2023, 24, 4547. [Google Scholar] [CrossRef]
- Ramesh, V.; Nair, D.; Zhang, S.X.; Hakim, F.; Kaushal, N.; Kayali, F.; Wang, Y.; Li, R.C.; Carreras, A.; Gozal, D. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-alpha pathway. J. Neuroinflamm. 2012, 9, 91. [Google Scholar] [CrossRef]
- de la Paz Ramirez, P.; Ordaz, G.; de la Paz Gonzales, R.; Perez, R.E.; Lopez, M.; Ortiz, R. Validation of portable electronic equipment (Accutrend((R)) Plus) to determine glucose, total cholesterol, and triglycerides in rats (Rattus) and dogs (Canis lupus familiaris). J. Adv. Vet. Anim. Res. 2023, 10, 57–63. [Google Scholar] [CrossRef]
- Dahan, T.; Nassar, S.; Yajuk, O.; Steinberg, E.; Benny, O.; Abudi, N.; Plaschkes, I.; Benyamini, H.; Gozal, D.; Abramovitch, R.; et al. Chronic Intermittent Hypoxia during Sleep Causes Browning of Interscapular Adipose Tissue Accompanied by Local Insulin Resistance in Mice. Int. J. Mol. Sci. 2022, 23, 15462. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2^(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [Google Scholar] [PubMed]
- Sen, S.; Domingues, C.C.; Rouphael, C.; Chou, C.; Kim, C.; Yadava, N. Genetic modification of human mesenchymal stem cells helps to reduce adiposity and improve glucose tolerance in an obese diabetic mouse model. Stem Cell Res. Ther. 2015, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Yosten, G.L.; Kolar, G.R. The Physiology of Proinsulin C-Peptide: Unanswered Questions and a Proposed Model. Physiology 2015, 30, 327–332. [Google Scholar] [CrossRef]
- Wahren, J.; Kallas, A.; Sima, A.A. The clinical potential of C-peptide replacement in type 1 diabetes. Diabetes 2012, 61, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Drager, L.F.; Togeiro, S.M.; Polotsky, V.Y.; Lorenzi-Filho, G. Obstructive sleep apnea: A cardiometabolic risk in obesity and the metabolic syndrome. J. Am. Coll. Cardiol. 2013, 62, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Reutrakul, S.; Mokhlesi, B. Obstructive Sleep Apnea and Diabetes: A State of the Art Review. Chest 2017, 152, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Benkirane, O.; Delwiche, B.; Mairesse, O.; Peigneux, P. Impact of Sleep Fragmentation on Cognition and Fatigue. Int. J. Environ. Res. Public Health 2022, 19, 15485. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and inflammation—Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; An, Y.A.; Scherer, P.E. Mitochondrial regulation and white adipose tissue homeostasis. Trends Cell Biol. 2022, 32, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Almendros, I.; Garcia-Rio, F. Sleep apnoea, insulin resistance and diabetes: The first step is in the fat. Eur. Respir. J. 2017, 49, 1700179. [Google Scholar] [CrossRef]
- Owino, S.; Buonfiglio, D.D.C.; Tchio, C.; Tosini, G. Melatonin Signaling a Key Regulator of Glucose Homeostasis and Energy Metabolism. Front. Endocrinol. 2019, 10, 488. [Google Scholar] [CrossRef]
- Cipolla-Neto, J.; Amaral, F.G.; Afeche, S.C.; Tan, D.X.; Reiter, R.J. Melatonin, energy metabolism, and obesity: A review. J. Pineal Res. 2014, 56, 371–381. [Google Scholar] [CrossRef]
- Fernandez Vazquez, G.; Reiter, R.J.; Agil, A. Melatonin increases brown adipose tissue mass and function in Zucker diabetic fatty rats: Implications for obesity control. J. Pineal Res. 2018, 64, e12472. [Google Scholar] [CrossRef]
- Goransson, O.; Kopietz, F.; Rider, M.H. Metabolic control by AMPK in white adipose tissue. Trends Endocrinol. Metab. 2023, 34, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, C.; Negri, M.; Patalano, R.; Amatrudo, F.; Monto, T.; Liccardi, A.; Graziadio, C.; Muscogiuri, G.; Pivonello, R.; Colao, A. The role of melatonin in the molecular mechanisms underlying metaflammation and infections in obesity: A narrative review. Obes. Rev. 2022, 23, e13390. [Google Scholar] [CrossRef] [PubMed]
- Um, J.H.; Pendergast, J.S.; Springer, D.A.; Foretz, M.; Viollet, B.; Brown, A.; Kim, M.K.; Yamazaki, S.; Chung, J.H. AMPK regulates circadian rhythms in a tissue- and isoform-specific manner. PLoS ONE 2011, 6, e18450. [Google Scholar] [CrossRef] [PubMed]
- Reutrakul, S.; Van Cauter, E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metabolism 2018, 84, 56–66. [Google Scholar] [CrossRef]
- Garaulet, M.; Qian, J.; Florez, J.C.; Arendt, J.; Saxena, R.; Scheer, F. Melatonin Effects on Glucose Metabolism: Time to Unlock the Controversy. Trends Endocrinol. Metab. 2020, 31, 192–204. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.H.; Lee, D.-B.; Yoon, D.-W.; Kim, J. Melatonin Improves Glucose Homeostasis and Insulin Sensitivity by Mitigating Inflammation and Activating AMPK Signaling in a Mouse Model of Sleep Fragmentation. Cells 2024, 13, 470. https://doi.org/10.3390/cells13060470
Hong SH, Lee D-B, Yoon D-W, Kim J. Melatonin Improves Glucose Homeostasis and Insulin Sensitivity by Mitigating Inflammation and Activating AMPK Signaling in a Mouse Model of Sleep Fragmentation. Cells. 2024; 13(6):470. https://doi.org/10.3390/cells13060470
Chicago/Turabian StyleHong, Seok Hyun, Da-Been Lee, Dae-Wui Yoon, and Jinkwan Kim. 2024. "Melatonin Improves Glucose Homeostasis and Insulin Sensitivity by Mitigating Inflammation and Activating AMPK Signaling in a Mouse Model of Sleep Fragmentation" Cells 13, no. 6: 470. https://doi.org/10.3390/cells13060470
APA StyleHong, S. H., Lee, D.-B., Yoon, D.-W., & Kim, J. (2024). Melatonin Improves Glucose Homeostasis and Insulin Sensitivity by Mitigating Inflammation and Activating AMPK Signaling in a Mouse Model of Sleep Fragmentation. Cells, 13(6), 470. https://doi.org/10.3390/cells13060470