
Alcohol Exposure Induces Nucleolar Stress and Apoptosis in Mouse Neural Stem Cells and Late-Term Fetal Brain

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Alcohol Exposure of Animals
2.3. Whole Transcriptome Analysis of Fetal Cortex
2.4. Immunofluorescence Staining and Imaging of Fetal Cortex
2.5. Primary Neural Stem Cell (NSC) Cultures
2.6. NSC Transfection Studies
2.7. Immunofluorescent Staining and Imaging of NSCs
2.8. Quantification of NSC Proliferation
2.9. Assessment of NSC Apoptosis
2.10. Western Blot Analysis
2.11. Quantitative PCR (qPCR)
2.12. Statistical Analysis
3. Results
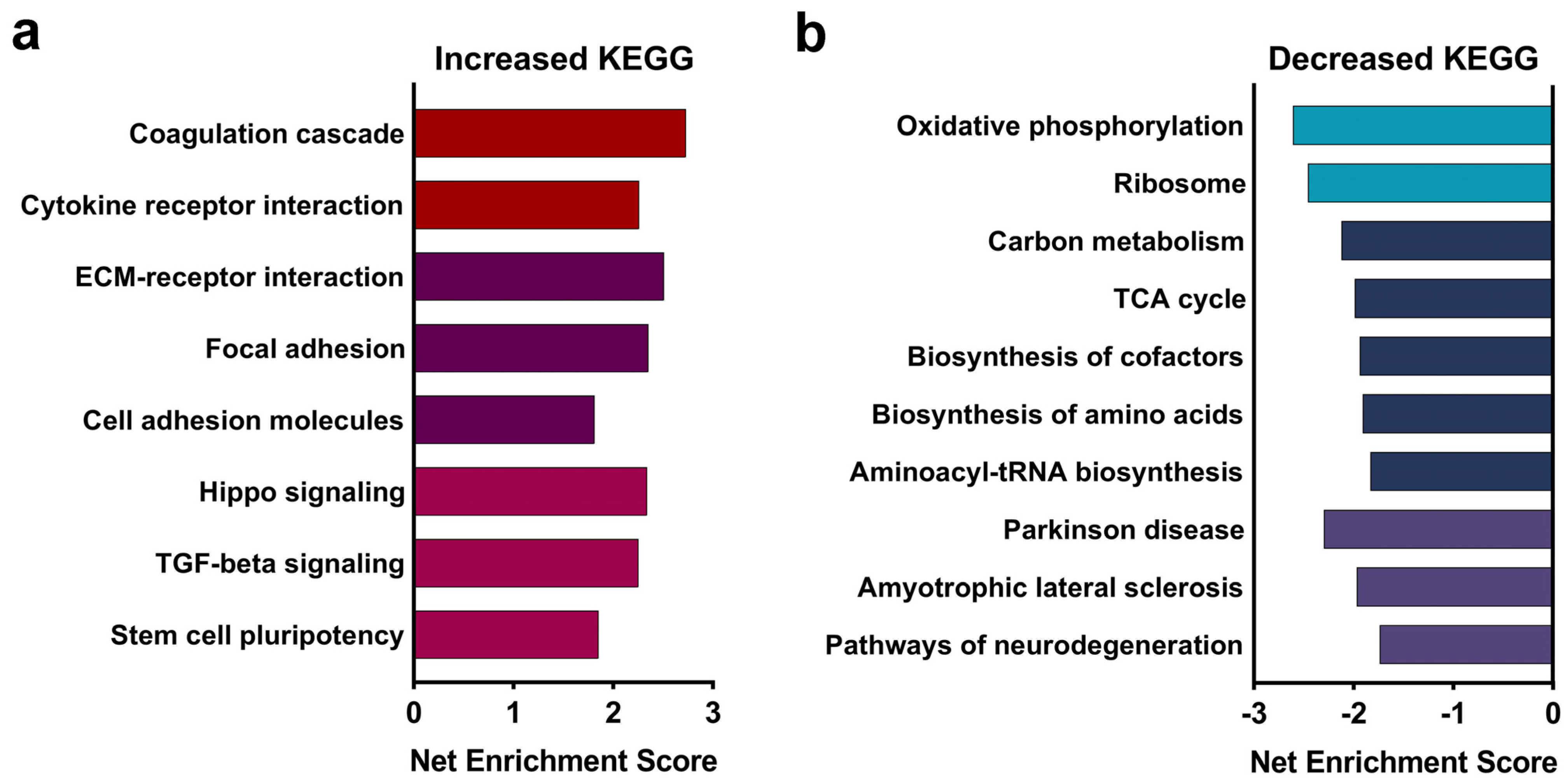
3.1. Alcohol Exposure Suppresses Pathways Associated with Ribosomes and Oxidative Phosphorylation in the E17.5 Fetal Brain
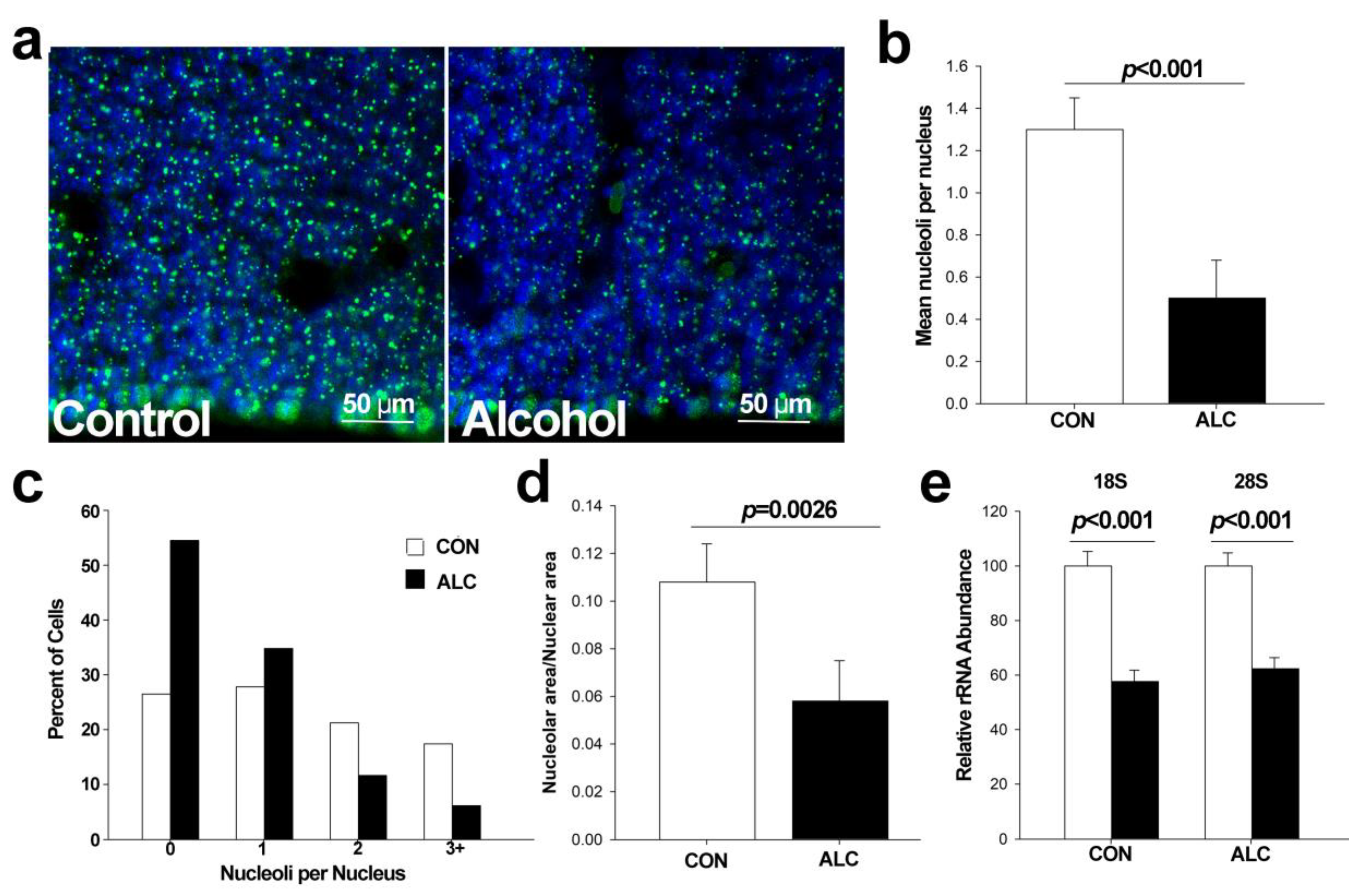
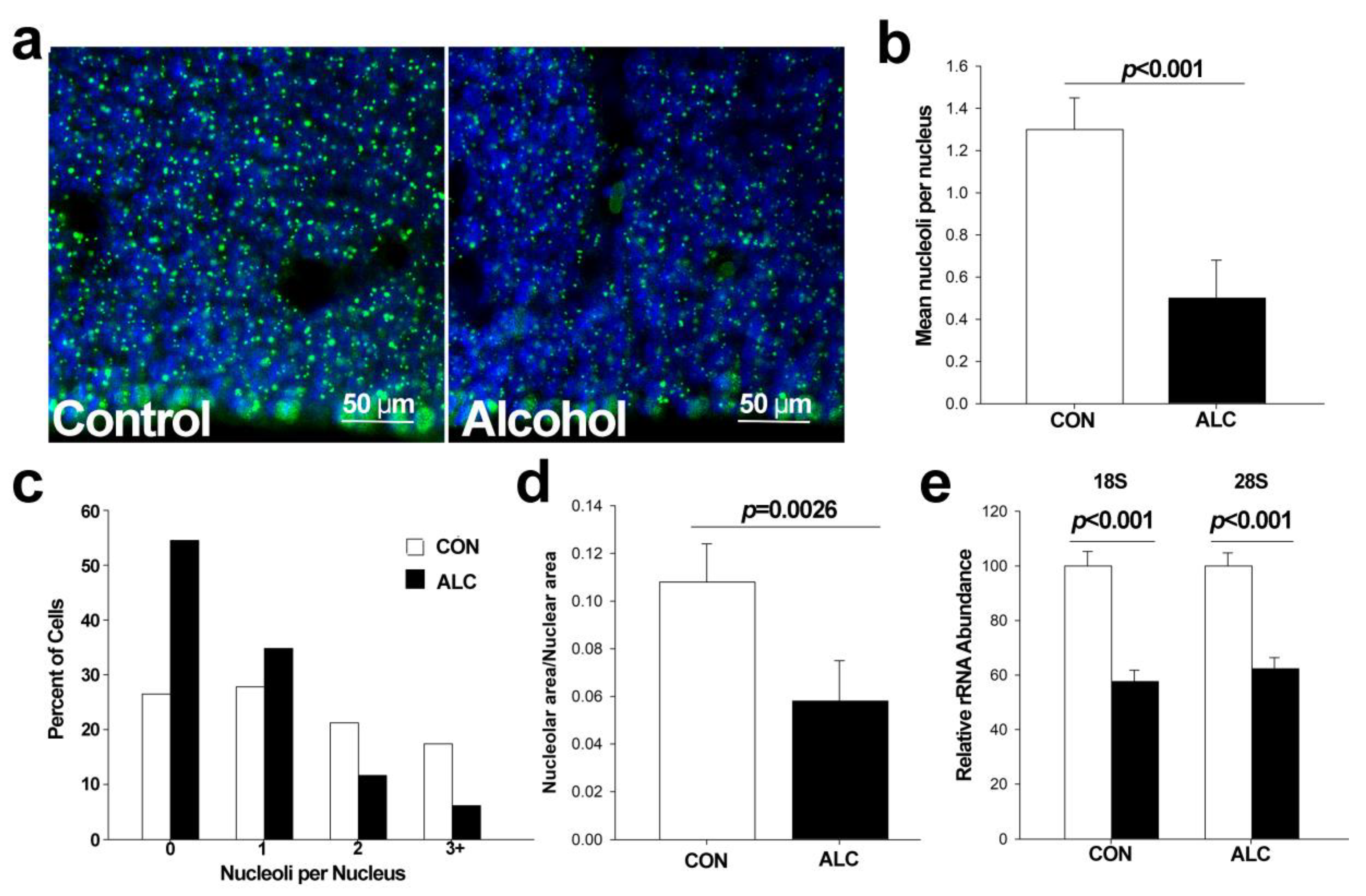
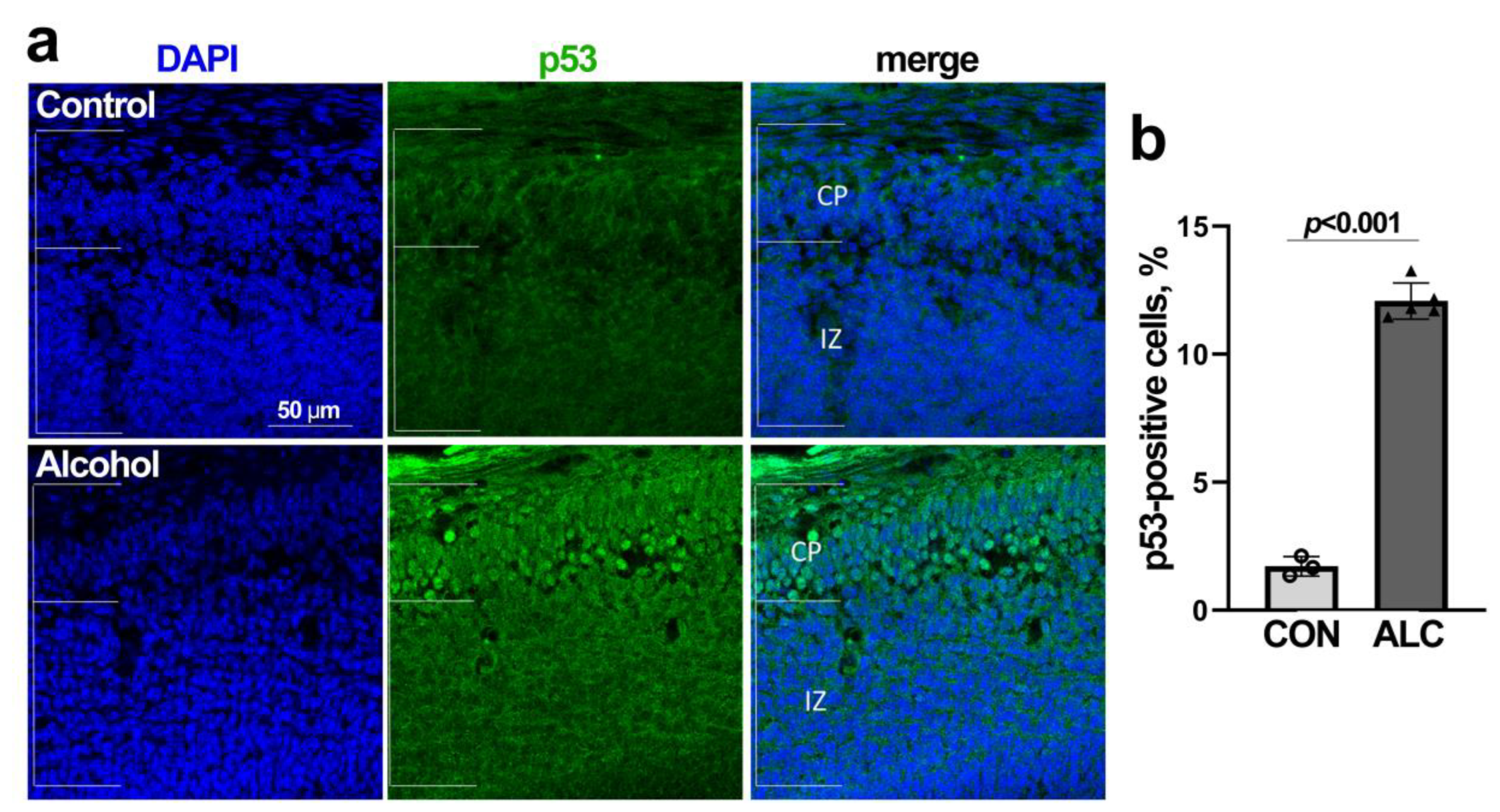
3.2. Alcohol Reduces Nucleoli Numbers in Fetal Cortex
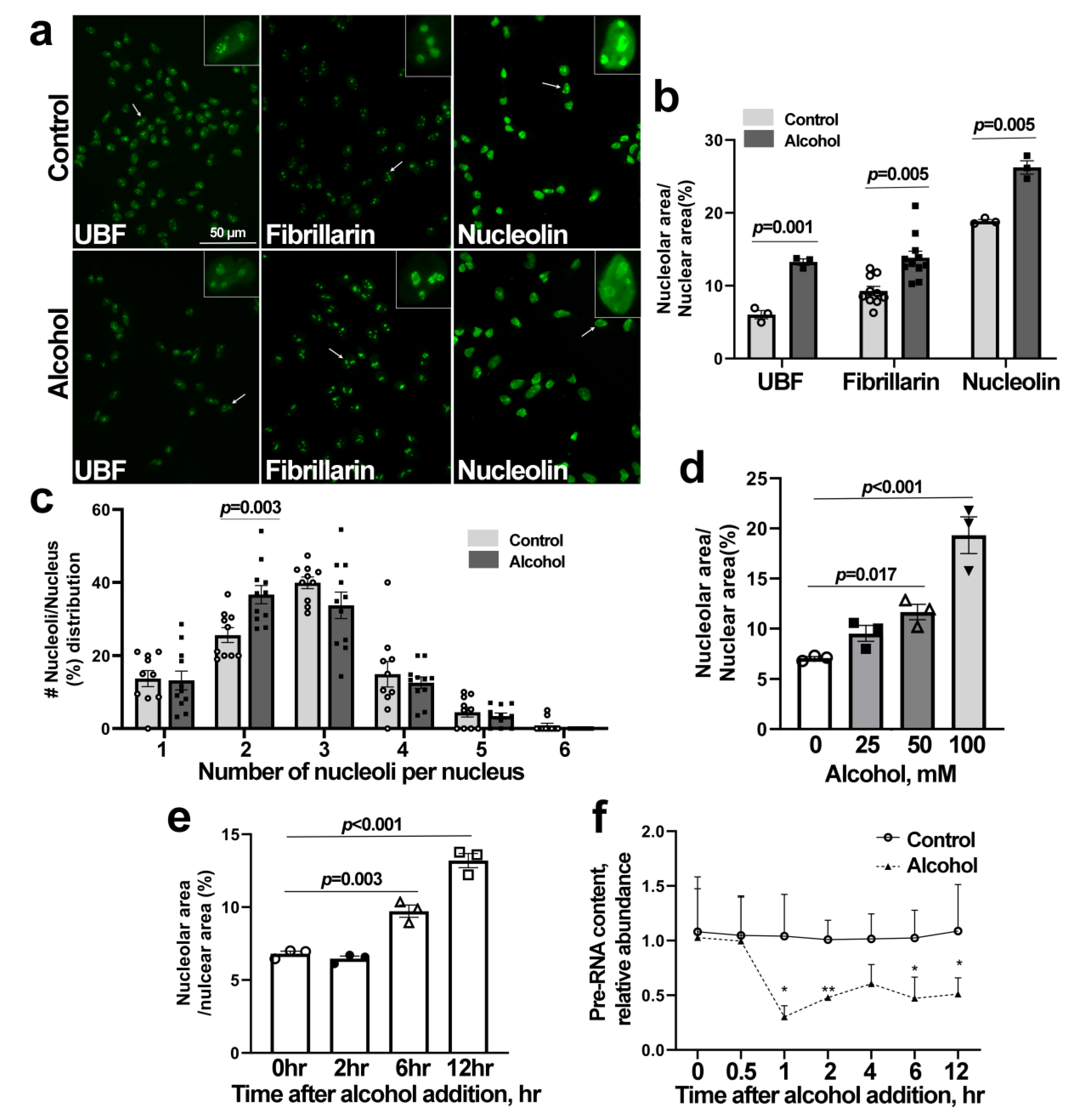
3.3. Alcohol Induces Nucleolar Stress in Cortical Neural Stem Cells (NSCs)
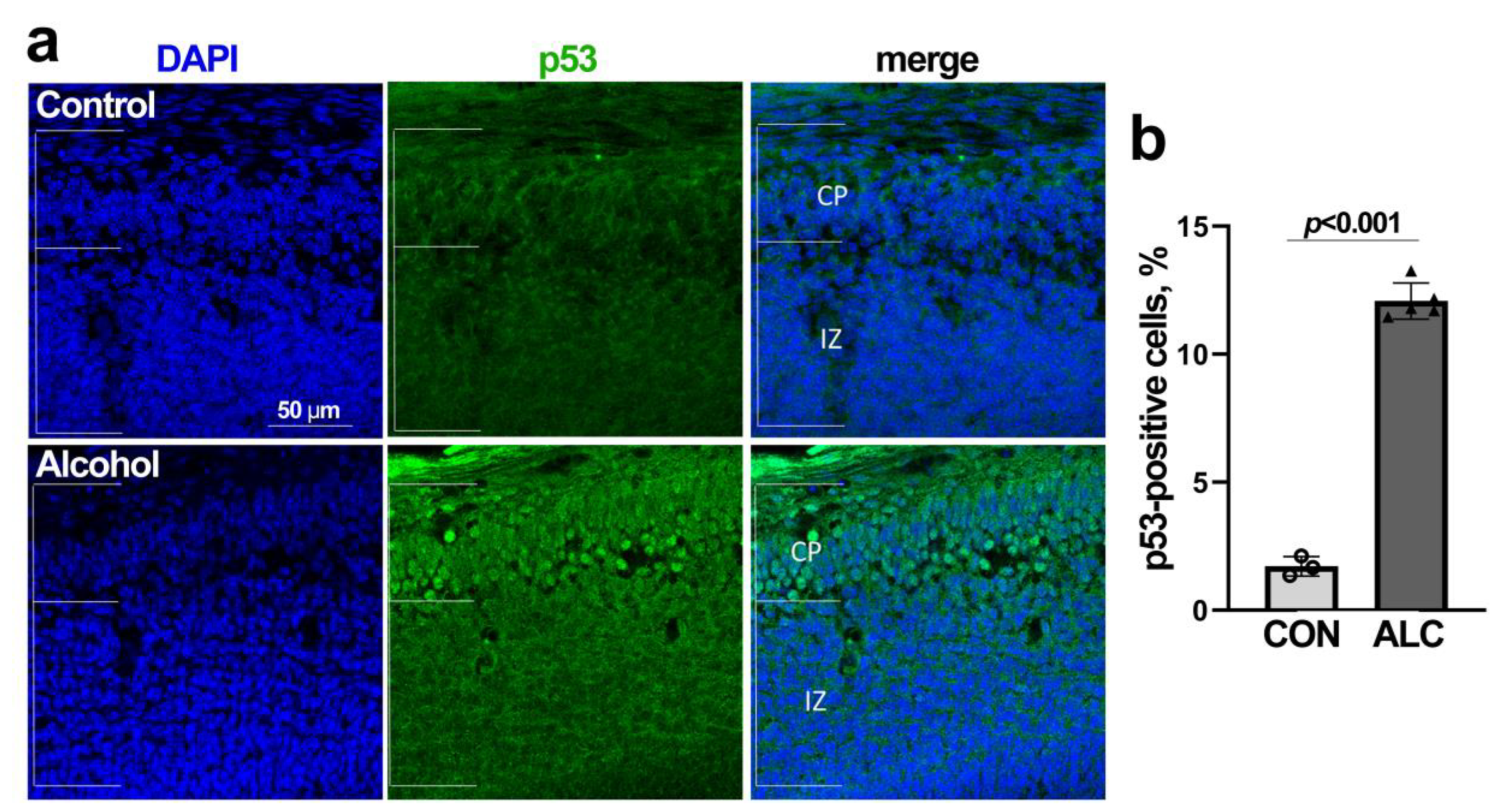
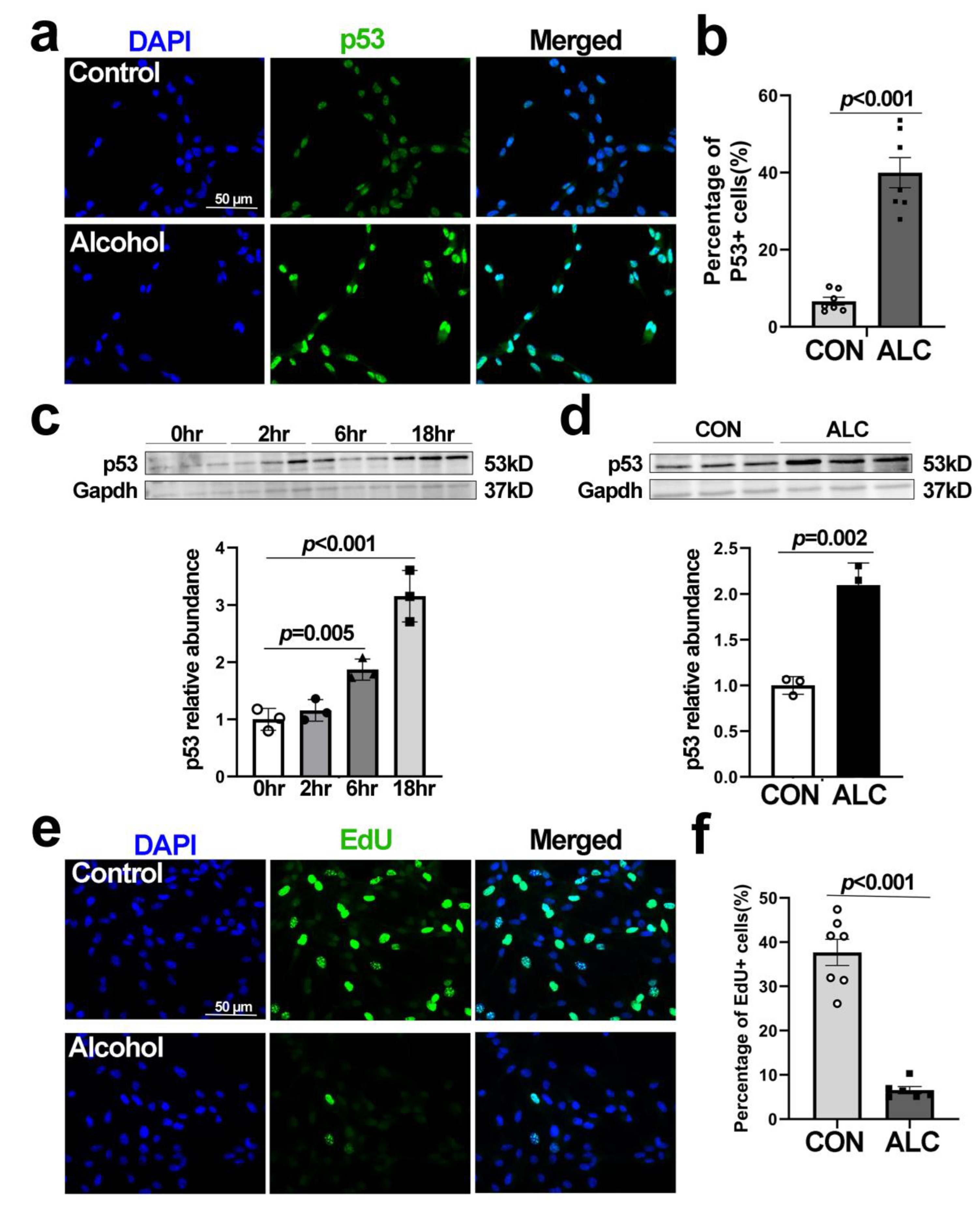
3.4. Alcohol-Induced Nucleolar Stress in NSCs Is Accompanied by Stabilization of Nuclear p53 and Cell Cycle Arrest
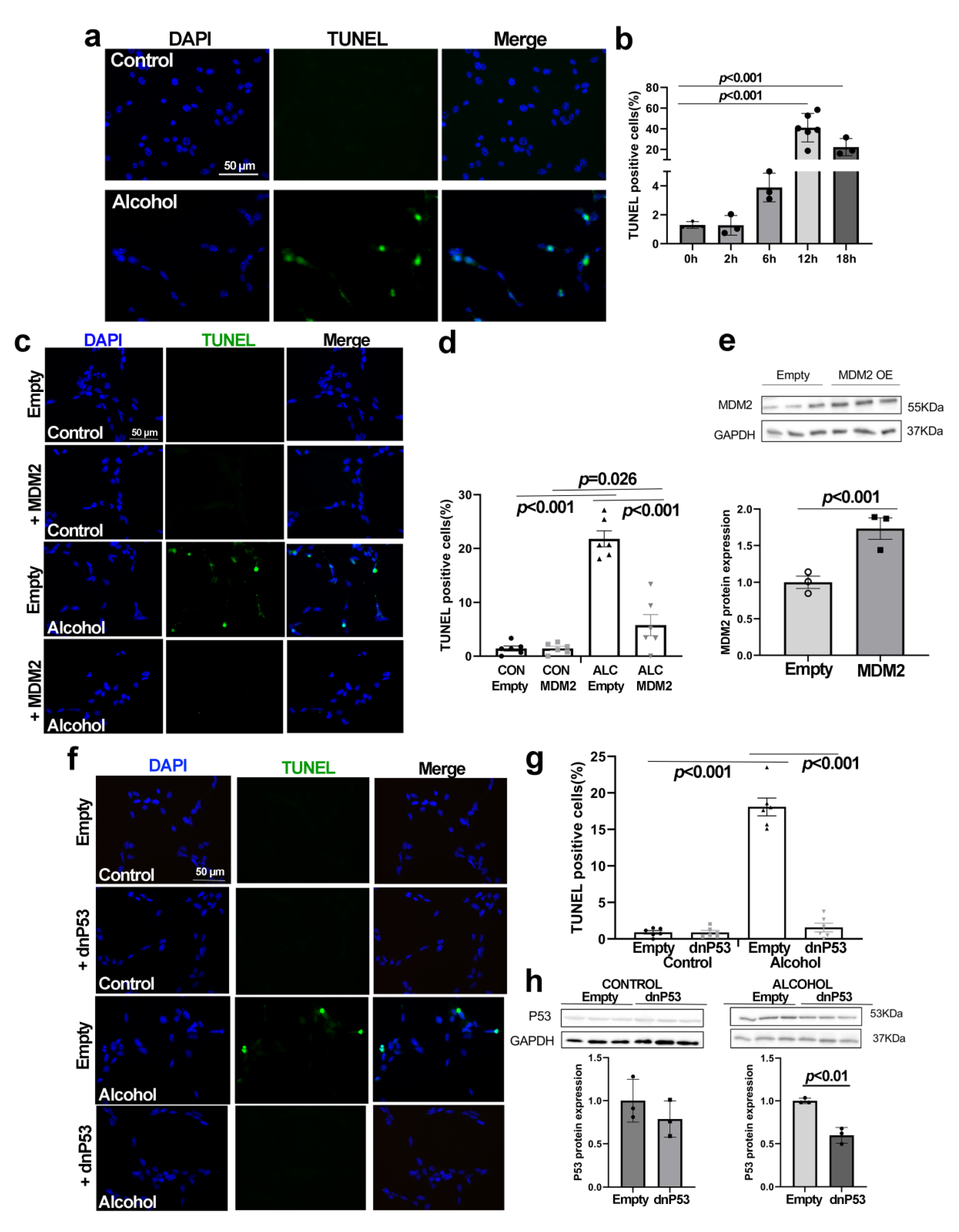
3.5. Alcohol Induces p53-Dependent Apoptosis in NSCs That Is Abrogated by MDM2
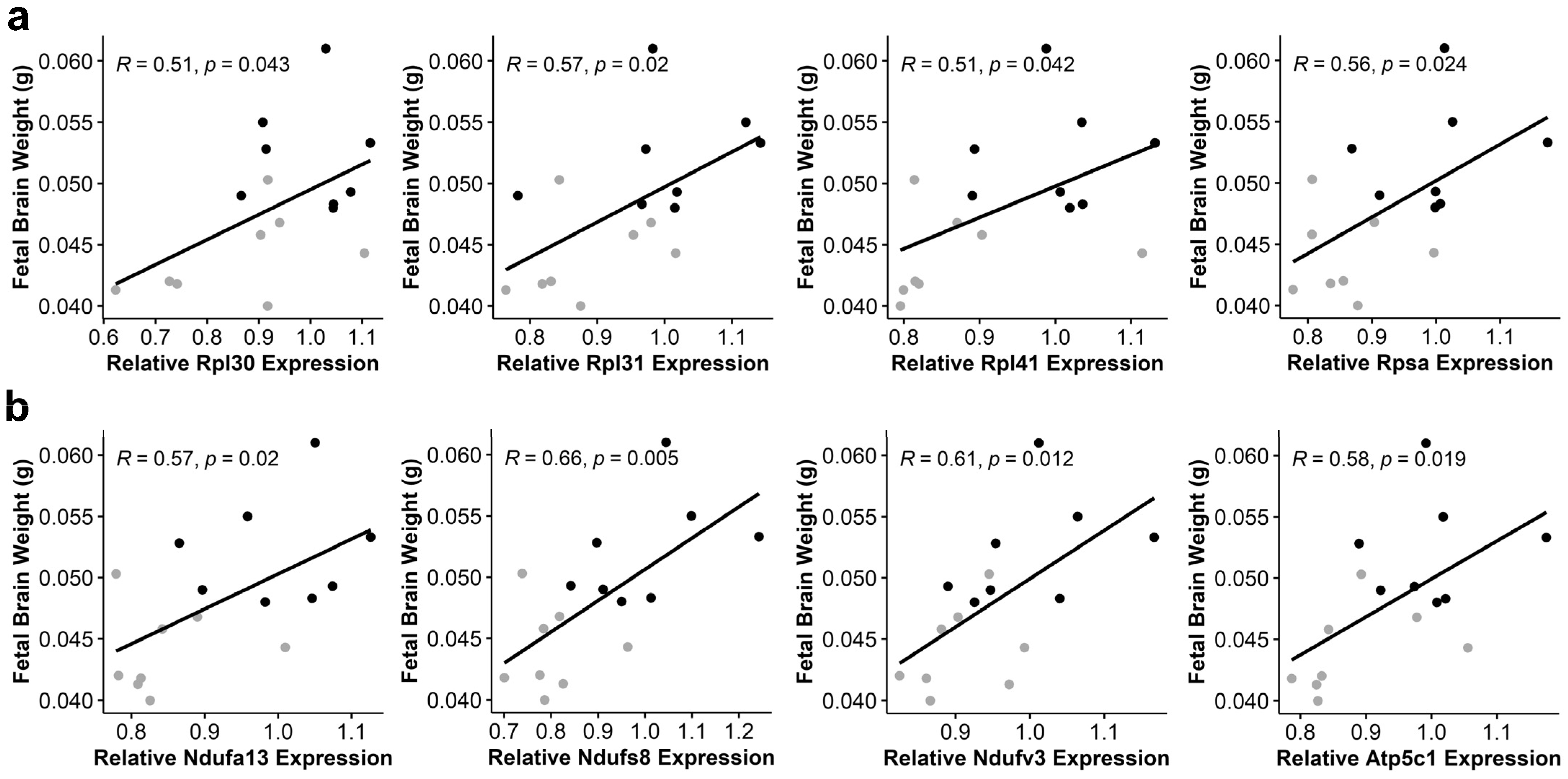
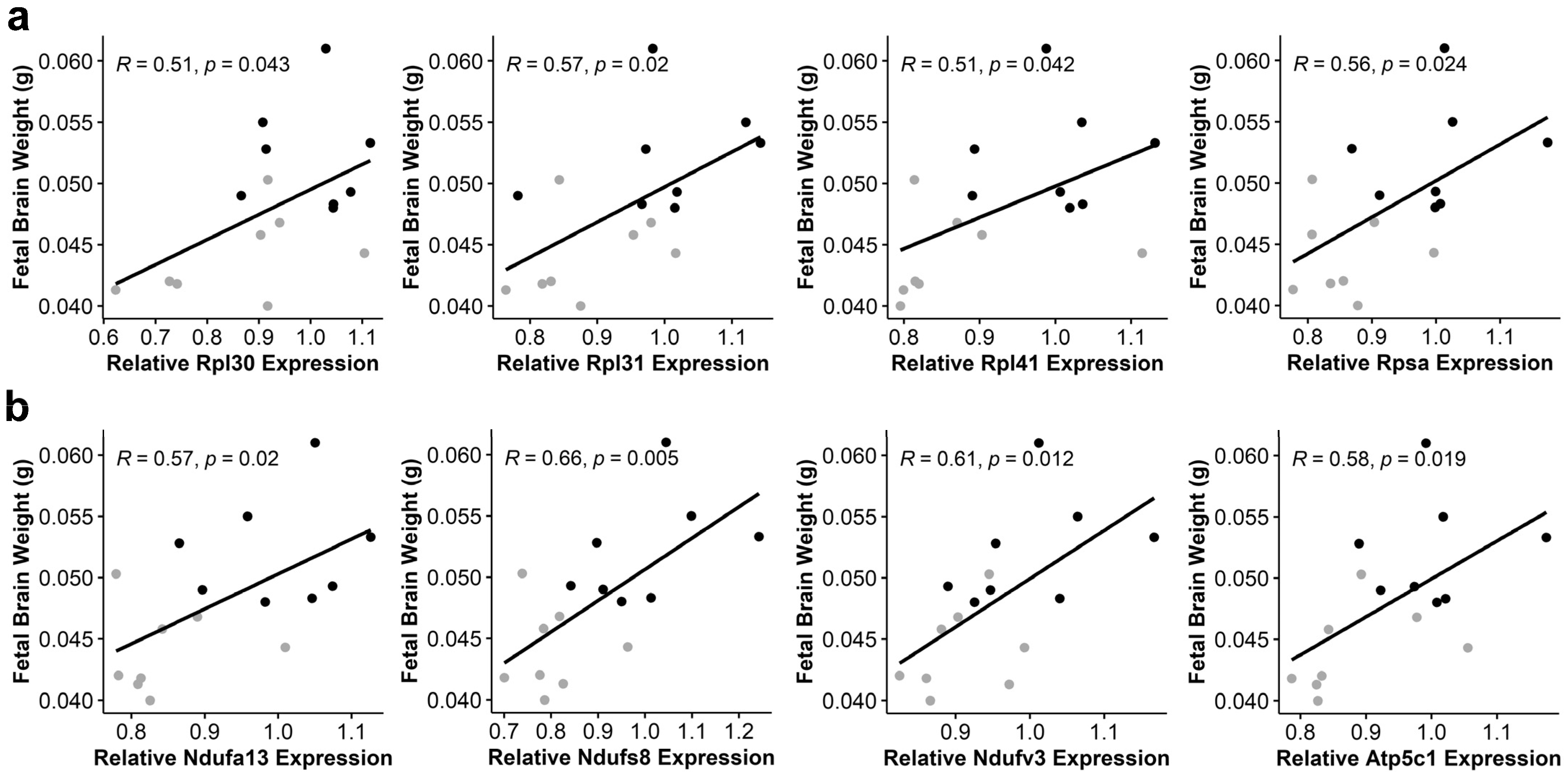
3.6. Alcohol-Induced Suppression of Ribosomal Proteins and Oxidative Phosphorylation Components Negatively Correlates with Fetal Brain Weight
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoyme, H.E.; Kalberg, W.O.; Elliott, A.J.; Blankenship, J.; Buckley, D.; Marais, A.-S.; Manning, M.A.; Robinson, L.K.; Adam, M.P.; Abdul-Rahman, O. Updated clinical guidelines for diagnosing fetal alcohol spectrum disorders. Pediatrics 2016, 138, e20154256. [Google Scholar] [CrossRef]
- May, P.A.; Chambers, C.D.; Kalberg, W.O.; Zellner, J.; Feldman, H.; Buckley, D.; Kopald, D.; Hasken, J.M.; Xu, R.; Honerkamp-Smith, G.; et al. Prevalence of fetal alcohol spectrum disorders in 4 US communities. JAMA 2018, 319, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Glass, L.; Moore, E.M.; Mattson, S.N. Current considerations for fetal alcohol spectrum disorders: Identification to intervention. Curr. Opin. Psychiatry 2023, 36, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Hyland, M.T.; Courchesne-Krak, N.S.; Bernes, G.A.; Wozniak, J.R.; Jones, K.L.; Del Campo, M.; Riley, E.P.; Mattson, S.N.; CIFASD. Results of a screening tool for fetal alcohol spectrum disorders are associated with neuropsychological and behavioral measures. Alcohol Clin. Exp. Res. 2023, 47, 1560–1569. [Google Scholar] [CrossRef]
- Bâ, A. Neurobiology of neuronal network alteration in intellectual disability related to fetal alcohol spectrum disorders. J. Beha Brain Sci. 2022, 12, 43–81. [Google Scholar] [CrossRef]
- Rubert, G.; Miñana, R.; Pascual, M.; Guerri, C. Ethanol exposure during embryogenesis decreases the radial glial progenitorpool and affects the generation of neurons and astrocytes. J. Neurosci. Res. 2006, 84, 483–496. [Google Scholar] [CrossRef]
- Miller, M.W.; Nowakowski, N.S. Effect of prenatal exposure to ethanol on the cell cycle kinetics and growth fraction in the proliferative zones of fetal rat cerebral cortex. Alcohol Clin. Exp. Res. 1991, 15, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Santillano, D.R.; Kumar, L.S.; Prock, T.L.; Camarillo, C.; Tingling, J.D.; Miranda, R.C. Ethanol induces cell-cycle activity and reduces stem cell diversity to alter both regenerative capacity and differentiation potential of cerebral cortical neuroepithelial precursors. BMC Neurosci. 2005, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R.; Vilardell, J.; Sohn, J.H. Economics of ribosome biosynthesis. Cold Spring Harb. Symp. Quant. Biol. 2001, 66, 567–574. [Google Scholar] [CrossRef]
- Dubois, M.-L.; Boisvert, F.-M. The Functional Nucleolus. In The Nucleolus: Structure and Function; Springer International Publishing: Cham, Switzerland, 2016; pp. 29–49. [Google Scholar]
- Lindstrom, M.S.; Bartek, J.; Maya-Mendoza, A. p53 at the crossroad of DNA replication and ribosome biogenesis stress pathways. Cell Death Differ. 2022, 29, 972–982. [Google Scholar] [CrossRef]
- Iadevaia, V.; Liu, R.; Proud, C.G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef]
- Jiao, L.; Liu, Y.; Yu, X.Y.; Pan, X.; Zhang, Y.; Tu, J.; Song, Y.H.; Li, Y. Ribosome biogenesis in disease: New players and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 15. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Conacci-Sorrell, M. Nucleolar stress: From development to cancer. Semin. Cell Dev. Biol. 2023, 136, 64–74. [Google Scholar] [CrossRef]
- Deisenroth, C.; Franklin, D.A.; Zhang, Y. The evolution of the ribosomal protein-MDM2-p53 pathway. Perspect. Med. 2016, 6, a026138. [Google Scholar] [CrossRef]
- Yelick, P.C.; Trainor, P.A. Ribosomopathies: Global process, tissue specific defects. Rare Dis. 2015, 3, e1025185. [Google Scholar] [CrossRef]
- Hetman, M.; Slomnicki, L.P. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem. 2019, 148, 325–347. [Google Scholar] [CrossRef]
- Venturi, G.; Montanaro, L. How altered ribosome production can cause or contribute to human disease: The spectrum of ribosomopathies. Cells 2020, 9, 2300. [Google Scholar] [CrossRef]
- Berres, M.E.; Garic, A.; Flentke, G.R.; Smith, S.M. Transcriptome profiling identifies ribosome biogenesis as a target of alcohol teratogenicity and vulnerability during early embryogenesis. PLoS ONE 2017, 12, e0169351. [Google Scholar] [CrossRef]
- Garic, A.; Berres, M.E.; Smith, S.M. High-throughput transcriptome sequencing identifies candidate genetic modifiers of vulnerability to fetal alcohol spectrum disorders. Alcohol Clin. Exp. Res. 2014, 38, 1874–1882. [Google Scholar] [CrossRef]
- Boschen, K.E.; Steensen, M.C.; Simon, J.M.; Parnell, S.E. Short-term transcriptomic changes in the mouse neural tube induced by an acute alcohol exposure. Alcohol 2023, 106, 1–9. [Google Scholar] [CrossRef]
- Downing, C.; Flink, S.; Florez-McClure, M.L.; Johnson, T.E.; Tabakoff, B.; Kechris, K.J. Gene expression changes in C57BL/6J and DBA/2J mice following prenatal alcohol exposure. Alcohol Clin. Exp. Res. 2012, 36, 1519–1529. [Google Scholar] [CrossRef]
- Flentke, G.R.; Baulch, J.W.; Berres, M.E.; Garic, A.; Smith, S.M. Alcohol-mediated calcium signals dysregulate pro-survival Snai2/PUMA/Bcl2 networks to promote p53-mediated apoptosis in avian neural crest progenitors. Birth Defects Res. 2019, 111, 686–699. [Google Scholar] [CrossRef]
- Fish, E.W.; Tucker, S.K.; Peterson, R.L.; Eberhart, J.K.; Parnell, S.E. Loss of tumor protein 53 protects against alcohol-induced facial malformations in mice and zebrafish. Alcohol Clin. Exp. Res. 2021, 45, 1965–1979. [Google Scholar] [CrossRef]
- Camargo Moreno, M.; Mooney, S.M.; Middleton, F.A. Heterogeneity of p53 dependent genomic responses following ethanol exposure in a developmental mouse model of fetal alcohol spectrum disorder. PLoS ONE 2017, 12, e0180873. [Google Scholar] [CrossRef]
- Mooney, S.M.; Pjetri, E.; Friday, W.B.; Smith, S.M. Growth and behavioral differences in a C57BL/6J mouse model of prenatal alcohol exposure. Alcohol 2021, 97, 51–57. [Google Scholar] [CrossRef]
- Smith, S.M.; Pjetri, E.; Friday, W.B.; Presswood, B.H.; Ricketts, D.K.; Walter, K.R.; Mooney, S.M. Aging-related behavioral, adiposity, and glucose impairments and their association following prenatal alcohol exposure in the C57BL/6J mouse. Nutrients 2022, 14, 1438. [Google Scholar] [CrossRef] [PubMed]
- Kwan, S.T.; Ricketts, D.K.; Presswood, B.H.; Smith, S.M.; Mooney, S.M. Prenatal choline supplementation during mouse pregnancy has differential effects in alcohol-exposed fetal organs. Alcohol Clin. Exp. Res. 2021, 45, 2471–2484. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Mandelboum, S.; Manber, Z.; Elroy-Stein, O.; Elkon, R. Recurrent functional misinterpretation of RNA-seq data caused by sample-specific gene length bias. PLoS Biol. 2019, 17, e3000481. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef]
- Rasband, W.; US National Institutes of Health. ImageJ, 2018; US National Institutes of Health: Bethesda, MD, USA, 2019.
- Camarillo, C.; Kumar, L.S.; Bake, S.; Sohrabji, F.; Miranda, R.C. Ethanol regulates angiogenic cytokines during neural development: Evidence from an in vitro model of mitogen-withdrawal–induced cerebral cortical neuroepithelial differentiation. Alcohol. Clin. Exp. Res. 2007, 31, 324–335. [Google Scholar] [CrossRef]
- Pinson, M.R.; Chung, D.D.; Mahnke, A.H.; Salem, N.A.; Osorio, D.; Nair, V.; Payne, E.A.; Del Real, J.J.; Cai, J.J.; Miranda, R.C. Gag-like proteins: Novel mediators of prenatal alcohol exposure in neural development. Alcohol. Clin. Exp. Res. 2022, 46, 556–569. [Google Scholar] [CrossRef]
- Kim, D.W.; Wu, N.; Kim, Y.C.; Cheng, P.F.; Basom, R.; Kim, D.; Dunn, C.T.; Lee, A.Y.; Kim, K.; Lee, C.S.; et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016, 30, 1289–1299. [Google Scholar] [CrossRef]
- Martynoga, B.; Drechsel, D.; Guillemot, F. Molecular control of neurogenesis: A view from the mammalian cerebral cortex. Cold Spring Harbor Perspect. Biol. 2012, 4, a008359. [Google Scholar] [CrossRef]
- Ochs, R.L.; Lischwe, M.A.; Spohn, W.H.; Busch, H. Fibrillarin: A new protein of the nucleolus identified by autoimmune sera. Biol. Cell 1985, 54, 123–133. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Marchetti, A.; Falcioni, R.; Soddu, S.; Mercurio, A.M. Activation of p53 function in carcinoma cells by the alpha6beta4 integrin. J. Biol. Chem. 1999, 274, 20733–20737. [Google Scholar] [CrossRef]
- Hicks, S.D.; Miller, M.W. Ethanol-induced DNA repair in neural stem cells is transforming growth factor β1-dependent. Exp. Neurol. 2019, 317, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Chittiboyina, S.; Bai, Y.; Abad, P.C.; Vidi, P.A.; Stauffacher, C.V.; Lelievre, S.A. The nuclear mitotic apparatus protein NuMA controls rDNA transcription and mediates the nucleolar stress response in a p53-independent manner. Nucleic Acids Res. 2017, 45, 11725–11742. [Google Scholar] [CrossRef]
- Miller, M.; Peter, A.; Wharton, S.; Wyllie, A. Proliferation and death of conditionally immortalized neural cells from murine neocortex: p53 alters the ability of neuron-like cells to re-enter the cell cycle. Brain Res. 2003, 965, 57–66. [Google Scholar] [CrossRef]
- Anthony, B.; Zhou, F.C.; Ogawa, T.; Goodlett, C.R.; Ruiz, J. Alcohol exposure alters cell cycle and apoptotic events during early neurulation. Alcohol Alcohol. 2008, 43, 261–273. [Google Scholar] [CrossRef]
- Miller, M.W. p53-Mediated Activities in NS-5 Neural Stem Cells: Effects of Ethanol. Alcohol. Clin. Exp. Res. 2019, 43, 655–667. [Google Scholar] [CrossRef]
- Yuan, F.; Yun, Y.; Fan, H.; Li, Y.; Lu, L.; Liu, J.; Feng, W.; Chen, S.Y. MicroRNA-135a protects against ethanol-induced apoptosis in neural crest cells and craniofacial defects in zebrafish by modulating the Siah1/p38/p53 pathway. Front. Cell Dev. Biol. 2020, 8, 583959. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, X.; Liu, J.; Feng, W.; Wu, X.; Chen, S.Y. Up-regulation of Siah1 by ethanol triggers apoptosis in neural crest cells through p38 MAPK-mediated activation of p53 signaling pathway. Arch. Toxicol. 2017, 91, 775–784. [Google Scholar] [CrossRef]
- Tran, T.D.; Cronise, K.; Marino, M.D.; Jenkins, W.J.; Kelly, S.J. Critical periods for the effects of alcohol exposure on brain weight, body weight, activity and investigation. Behav. Brain Res. 2000, 116, 99–110. [Google Scholar] [CrossRef]
- Zhou, F.C.; Sari, Y.; Powrozek, T.A.; Spong, C.Y. A neuroprotective peptide antagonizes fetal alcohol exposure-compromised brain growth. J. Mol. Neurosci. 2004, 24, 189–199. [Google Scholar] [CrossRef]
- Abbott, C.W.; Kozanian, O.O.; Kanaan, J.; Wendel, K.M.; Huffman, K.J. The impact of prenatal ethanol exposure on neuroanatomical and behavioral development in mice. Alcohol. Clin. Exp. Res. 2016, 40, 122–133. [Google Scholar] [CrossRef]
- Miller, M.W. Effect of pre- or postnatal exposure to ethanol on the total number of neurons in the principal sensory nucleus of the trigeminal nerve: Cell proliferation and neuronal death. Alcohol. Clin. Exp. Res. 1995, 19, 1359–1363. [Google Scholar]
- Parlato, R.; Kreiner, G.; Erdmann, G.; Rieker, C.; Stotz, S.; Savenkova, E.; Berger, S.; Grummt, I.; Schutz, G. Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J. Neurosci. 2008, 28, 12759–12764. [Google Scholar] [CrossRef]
- Farooq, M.; Lindbaek, L.; Krogh, N.; Doganli, C.; Keller, C.; Monnich, M.; Goncalves, A.B.; Sakthivel, S.; Mang, Y.; Fatima, A.; et al. RRP7A links primary microcephaly to dysfunction of ribosome biogenesis, resorption of primary cilia, and neurogenesis. Nat. Commun. 2020, 11, 5816. [Google Scholar] [CrossRef]
- Kang, H.; Shin, J.H. Repression of rRNA transcription by PARIS contributes to Parkinson’s disease. Neurobiol. Dis. 2015, 73, 220–228. [Google Scholar] [CrossRef]
- Ding, Q.; Markesbery, W.R.; Chen, Q.; Li, F.; Keller, J.N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005, 25, 9171–9175. [Google Scholar] [CrossRef]
- Slomnicki, L.P.; Malinowska, A.; Kistowski, M.; Palusinski, A.; Zheng, J.J.; Sepp, M.; Timmusk, T.; Dadlez, M.; Hetman, M. Nucleolar Enrichment of Brain Proteins with Critical Roles in Human Neurodevelopment. Mol. Cell. Proteom. 2016, 15, 2055–2075. [Google Scholar] [CrossRef]
- Cullen, C.L.; Burne, T.H.; Lavidis, N.A.; Moritz, K.M. Low dose prenatal ethanol exposure induces anxiety-like behaviour and alters dendritic morphology in the basolateral amygdala of rat offspring. PLoS ONE 2013, 8, e54924. [Google Scholar] [CrossRef]
- Louth, E.L.; Luctkar, H.D.; Heney, K.A.; Bailey, C.D. Developmental ethanol exposure alters the morphology of mouse prefrontal neurons in a layer-specific manner. Brain Res. 2018, 1678, 94–105. [Google Scholar] [CrossRef]
- Li, H.L.; Wu, S.; Rottenberg, H. Alcohol inhibits the depolarization-induced stimulation of oxidative phosphorylation in synaptosomes. J. Neurochem. 1996, 66, 1691–1697. [Google Scholar] [CrossRef]
- Chang, R.C.; Thomas, K.N.; Mehta, N.A.; Veazey, K.J.; Parnell, S.E.; Golding, M.C. Programmed suppression of oxidative phosphorylation and mitochondrial function by gestational alcohol exposure correlate with widespread increases in H3K9me2 that do not suppress transcription. Epigenet. Chromatin 2021, 14, 27. [Google Scholar] [CrossRef]
- Arzua, T.; Yan, Y.; Jiang, C.; Logan, S.; Allison, R.L.; Wells, C.; Kumar, S.N.; Schafer, R.; Bai, X. Modeling alcohol-induced neurotoxicity using human induced pluripotent stem cell-derived three-dimensional cerebral organoids. Transl. Psychiatry 2020, 10, 347. [Google Scholar] [CrossRef]
- Chen, G.; Ke, Z.; Xu, M.; Liao, M.; Wang, X.; Qi, Y.; Zhang, T.; Frank, J.A.; Bower, K.A.; Shi, X.; et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012, 8, 1577–1589. [Google Scholar] [CrossRef]
- Hwang, C.J.; Kim, Y.E.; Son, D.J.; Park, M.H.; Choi, D.Y.; Park, P.H.; Hellstrom, M.; Han, S.B.; Oh, K.W.; Park, E.K.; et al. Parkin deficiency exacerbate ethanol-induced dopaminergic neurodegeneration by P38 pathway dependent inhibition of autophagy and mitochondrial function. Redox Biol. 2017, 11, 456–468. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Tseng, A.; Jensen, M.B.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75 (Suppl. S1), 8–18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | KEGG ID | Enrichment Score | # Significant/ Total Genes 1 | Padj | Gene Name |
|---|---|---|---|---|---|
| Up-Regulated Genes | |||||
| Cytokine–cytokine receptor interaction | mmu04060 | 2.26 | 15/35 | 0.0026 | Acvr1c, Bmp5, Bmp6, Bmp7, Bmpr2, Csf1, Csf1r, Cxcl12, Gdf11, Il17ra, Il6ra, Inha, Lepr, Tgfb3, Tgfbr2 |
| Complement and coagulation cascades | mmu04610 | 2.73 | 13/17 | 0.0026 | A2m, C3, Cd46, Cfb, Cfh, F13a1, Fga, Fgb, Fgg, Serpind1, Tfpi, Thbd, Vwf |
| Hippo signaling pathway | mmu04390 | 2.34 | 28/53 | 0.0026 | Afp, Amot, Apc, Axin2, Bmp5, Fmp6, Bmp7, Bmpr2, Cdh1, Dlg1, Dvl3, Fzd1, Fzd3, Fzd5, Fzd7, Fzd8, Gsk3b, Lats2, Mob1b, Smad3, Tcf7l1, Tead1, Tgfb3, Tgfbr2, Trp73, Wnt2b, Wwtr1, Yap1 |
| Focal adhesion | mmu04510 | 2.35 | 37/48 | 0.0026 | Col1a1, Col1a2, Col2a1, Col4a1, Col4a2, Col4a5, Col4a6, Col6a1, Col6a2, Col6a3, Flnb, Flnc, Fn1, Igf1r, Itga1, Itga11, Itga2, Itga5, Itgb3, Kdr, Lama1, Lama2, Lamb1, Lamb2, Lamc1, Lamc3, Mylk, Pdgfrb, Pxn, Reln, Rock1, Thbs2, Thbs3, Tnr, Vav3, Vegfa, Vwf |
| Down-Regulated Genes | |||||
| Oxidative phosphorylation | mmu00190 | −2.61 | 63/69 | 0.0026 | Atp5a1, Atp5b, Atp5c1, Atp5f1, Atp5g1, Atp5g3, Atp5j2, Atp5k, Atp5o, Atp6v0d1, Atp6v0e2, Atp6v1b2, Atp6v1f, Atp6v1h, Cox10, Cox11, Cox17, Cox5a, Cox5b, Cox6a1, Cox6b1, Cox7a2, Cox7a2l, Cox7b, Cox8a, Cyc1, Ndufa10, Ndufa13, Ndufa2, Ndufa3, Ndufa6, Ndufa7, Ndufa8, Ndufa9, Ndufb10, Ndufb11, Ndufb2, Ndufb4, Ndufb5, Ndufb6, Ndufb7, Ndufb8, Ndufb9, Ndufc1, Ndufc2, Ndufs4, Ndufs6, Ndufs7, Ndufs8, Ndufv1, Ndufv2, Ndufv3, Ppa1, Ppa2, Sdhb, Sdhc, Sdhd, Uqcr10, Uqcr11, Uqcrc1, Uqcrc2, Uqcrfs1, Uqcrq |
| Ribosome | mmu03010 | −2.46 | 74/94 | 0.0026 | Mrpl10, Mrpl11, Mrpl12, Mrpl13, Mrpl14, Mrpl15, Mrpl18, Mrpl19, Mrpl2, Mrpl20, Mrpl22, Mrpl27, Mrpl28, Mrpl3, Mrpl33, Mrpl34, Mrpl9, Mrps10, Mrps11, Mrps12, Mrps17, Mrps18a, Mrps21, Mrps6, Mrps7, Rpl3, Rpl6, Rpl8, Rpl11, Rpl12, Rpl13, Rpl13a, Rpl14, Rpl15, Rpl18, Rpl22l1, Rpl27, Rpl27a, Rpl28, Rpl29, Rpl30, Rpl31, Rpl35, Rpl36, Rpl36a, Rpl37, Rpl38, Rpl39, Rpl41, Rplp0, Rplp1, Rps2, Rps3, Rps5, Rps6, Rps7, Rps8, Rps9, Rps10, Rps13, Rps15, Rps16, Rps17, Rps18, Rps20, Rps21, Rps23, Rps24, Rps27a, Rps28, Rps29, Rpsa |
| Pathways of Neurodegeneration | mmu05022 | −1.74 | 93/143 | 0.0026 | Actr10, Actr1a, Atf4, Atp5a1, Atp5b, Atp5c1, Atp5f1, Atp5g1, Atp5h, Atp5o, Bad, Cox4i1, Cox5a, Cox5b, Cox6a1, Cox6b1, Cox7a2, Cox7a2l, Cox7b, Cox8a, Cyc1, Cycs, Daxx, Dctn3, Gabarap, Gpx1, Hsd17b10, Ift57, Ndufa10, Ndufa3, Ndufa2, Ndufa3, Ndufa6, Ndufa7, Ndufa8, Ndufa9, Ndufc1, Ndufc2, Ndufs2, Ndufs4, Ndufs6, Ndufs7, Ndufs8, Ndufv1, Ndufv2, Ndufv3, Park7, Ppif, Psma6, Psma7, Psmb1, Psmb4, Psmb5, Psmb6, Psmb7, Psmc2, Psmc3, Psmc4, Psmc5, Psmd1, Psmd6, Psmd8, Psmd9, Rab8a, Rps27a, Sdhb, Sdhc, Sdhd, Septin5, Sigmar1, Sqstm1, Tomm40, Tom40l, Trap1, Tuba1c, Tubb4b, Ubb, Uqcr10, Uqcr11, Uqcrc1, Uqcrc2, Uqcrfs1, Uqcrq, Vdac3, Xbp1 |
| Gene | R | p-Value |
|---|---|---|
| Atp5c1 | 0.58 | 0.019 |
| Atp5K1 | 0.56 | 0.023 |
| Atp6v0e2 | 0.55 | 0.027 |
| Ndufa13 | 0.57 | 0.020 |
| Ndufb2 | 0.53 | 0.037 |
| Ndufb11 | 0.53 | 0.037 |
| Ndufs4 | 0.51 | 0.045 |
| Ndufs8 | 0.66 | 0.005 |
| Ndufv3 | 0.61 | 0.012 |
| Ppa2 | 0.53 | 0.033 |
| Uqcrq | 0.54 | 0.032 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Flentke, G.R.; Rivera, O.C.; Saini, N.; Mooney, S.M.; Smith, S.M. Alcohol Exposure Induces Nucleolar Stress and Apoptosis in Mouse Neural Stem Cells and Late-Term Fetal Brain. Cells 2024, 13, 440. https://doi.org/10.3390/cells13050440
Huang Y, Flentke GR, Rivera OC, Saini N, Mooney SM, Smith SM. Alcohol Exposure Induces Nucleolar Stress and Apoptosis in Mouse Neural Stem Cells and Late-Term Fetal Brain. Cells. 2024; 13(5):440. https://doi.org/10.3390/cells13050440
Chicago/Turabian StyleHuang, Yanping, George R. Flentke, Olivia C. Rivera, Nipun Saini, Sandra M. Mooney, and Susan M. Smith. 2024. "Alcohol Exposure Induces Nucleolar Stress and Apoptosis in Mouse Neural Stem Cells and Late-Term Fetal Brain" Cells 13, no. 5: 440. https://doi.org/10.3390/cells13050440
APA StyleHuang, Y., Flentke, G. R., Rivera, O. C., Saini, N., Mooney, S. M., & Smith, S. M. (2024). Alcohol Exposure Induces Nucleolar Stress and Apoptosis in Mouse Neural Stem Cells and Late-Term Fetal Brain. Cells, 13(5), 440. https://doi.org/10.3390/cells13050440