
Modulations of Homeostatic ACE2, CD147, GRP78 Pathways Correlate with Vascular and Endothelial Performance Markers during Pulmonary SARS-CoV-2 Infection


 , , , ,
, , , ,  and
and 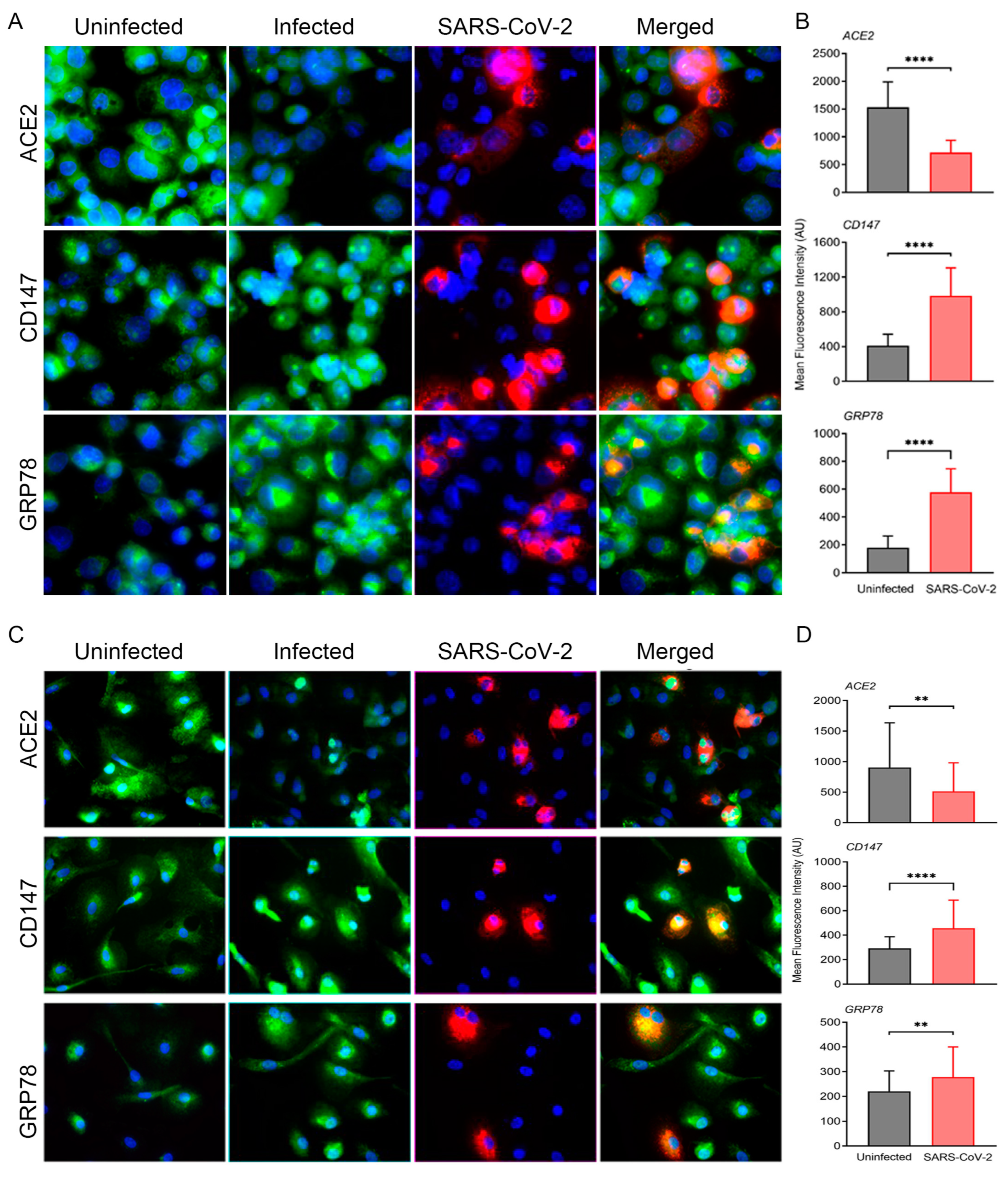{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Propagation of SARS-CoV-2 and In Vitro Infection
2.3. RNA Isolation and RT-qPCR
2.4. Hamster Infection and Sample Collection
2.5. Virus Infectivity Assays on Tissues
2.6. Histopathology Analysis
2.7. Single-Molecule Fluorescence In-Situ Hybridization (smFISH) Probes Synthesis
2.8. Processing of SARS-CoV-2-Infected THP-1 Cells for Imaging
2.9. Processing of Formalin-Fixed Paraffin-Embedded (FFPE) Tissues
2.10. Immunohistochemistry
2.11. Hybridization and smFISH Staining
2.12. Image Acquisition
2.13. Statistical Analysis
2.14. Ethics Statement
3. Results
3.1. ACE2-Dependent SARS-CoV-2 Infection of Cultured Human Macrophages Modulates the Expression of Key Homeostasis Pathways
3.2. SARS-CoV-2 Infected Hamsters Show Pathophysiology and Altered Homeostasis Resembling That of COVID-19 Patients Undergoing Proinflammatory and Prothrombotic Shifts
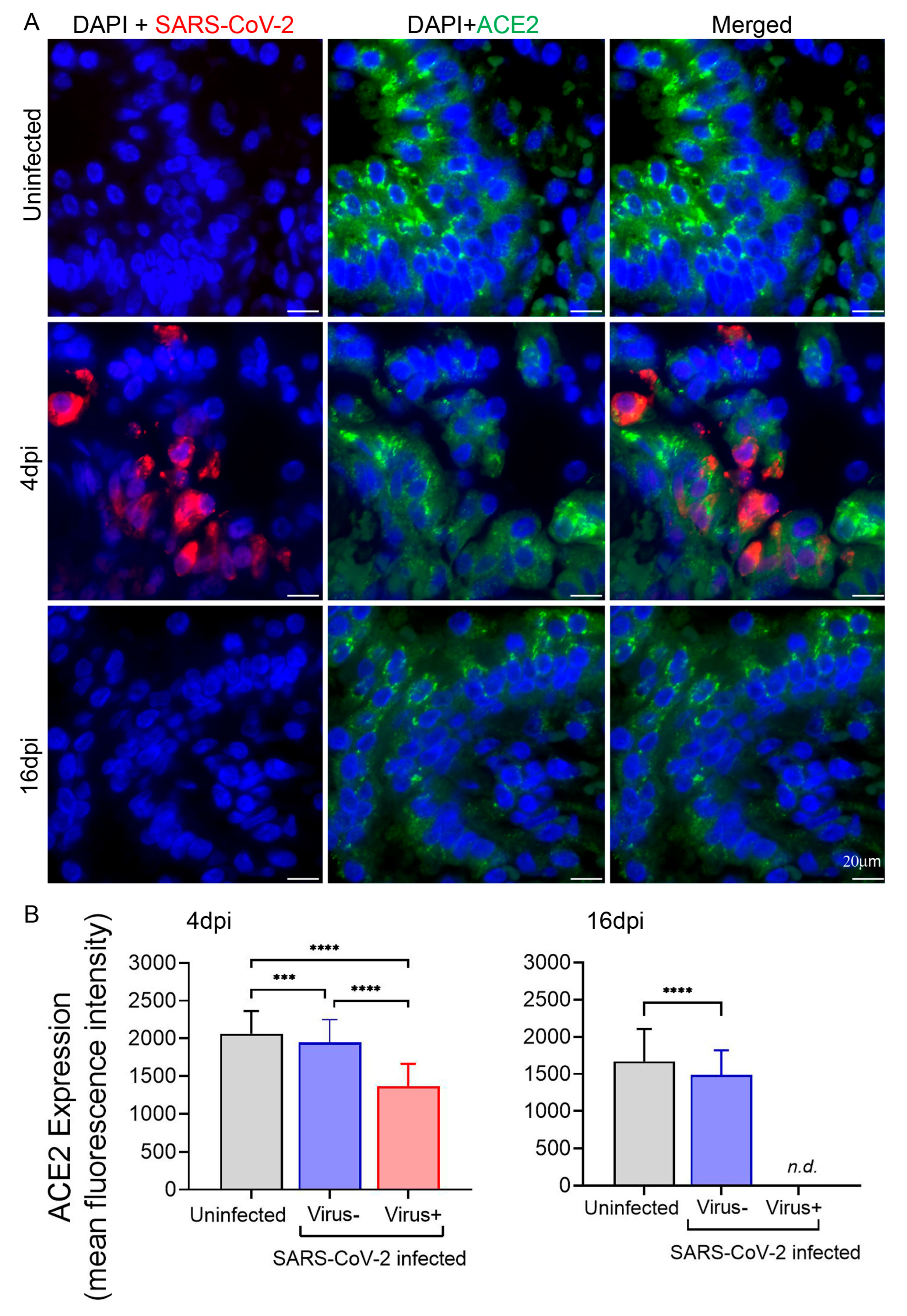
3.3. Expression of ACE2 in Hamster Lungs following SARS-CoV-2 Infection Is Downregulated in the Cells Harboring the Virus
3.4. SARS-CoV-2 Infection Upregulates Expression of CD147 and GRP78 in Hamster Lung Tissues
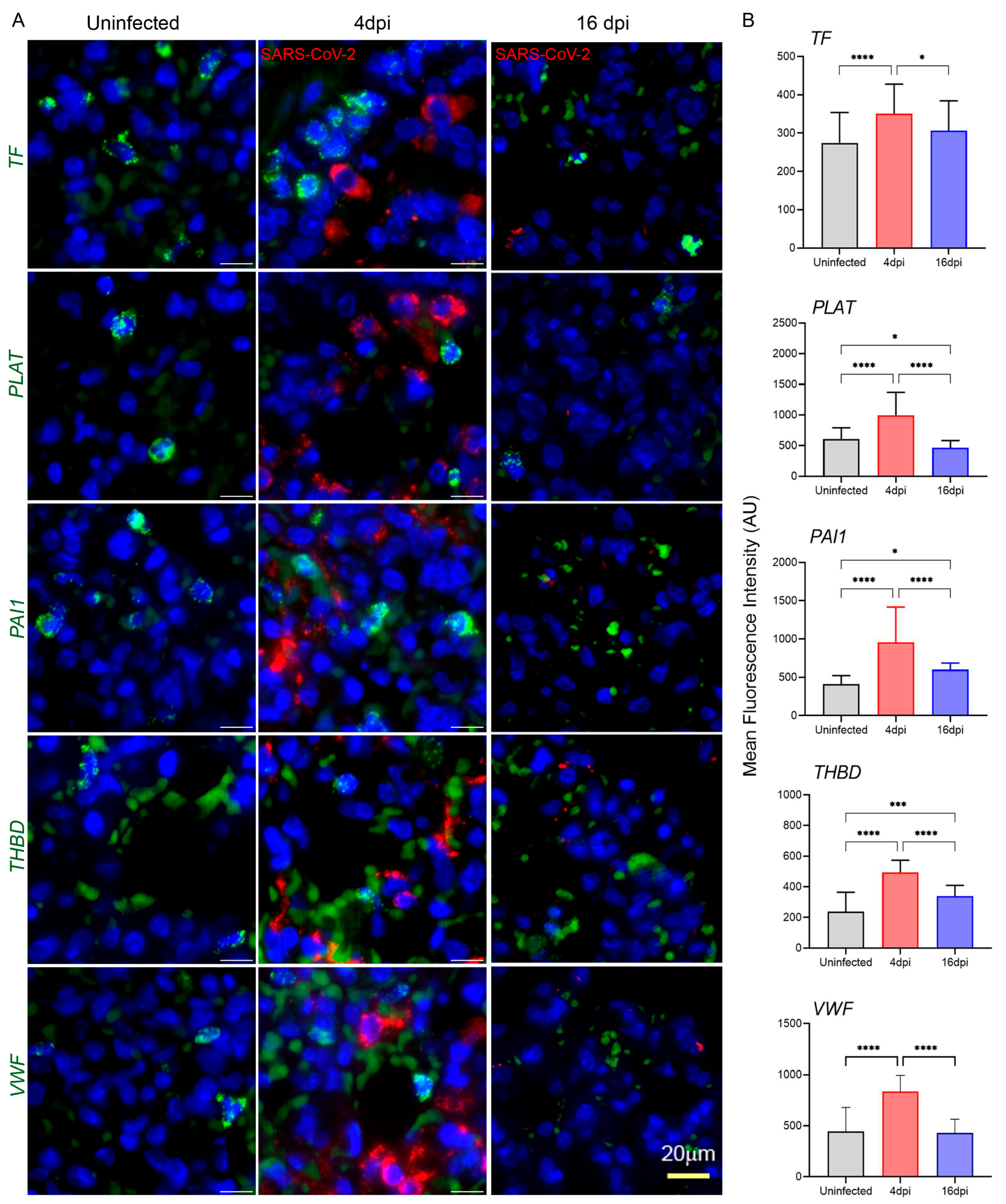
3.5. SARS-CoV-2 Infection Upregulates the Expression of Coagulation Cascade Markers That Reveal the Prothrombotic Shift in Hamster Lungs
3.6. SARS-CoV-2 Upregulates Markers of Endothelial Dysfunction in Infected Hamster Lungs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qing, E.; Gallagher, T. SARS Coronavirus Redux. Trends Immunol. 2020, 41, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Li, H.; Cao, B. A Novel Coronavirus (COVID-19) Outbreak: A Call for Action. Chest 2020, 157, e99–e101. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Garcia, L.F. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.K.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021, 40, e107826. [Google Scholar] [CrossRef]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef]
- Birnhuber, A.; Fliesser, E.; Gorkiewicz, G.; Zacharias, M.; Seeliger, B.; David, S.; Welte, T.; Schmidt, J.; Olschewski, H.; Wygrecka, M.; et al. Between inflammation and thrombosis: Endothelial cells in COVID-19. Eur. Respir. J. 2021, 58, 2100377. [Google Scholar] [CrossRef] [PubMed]
- Duhailib, Z.A.; Oczkowski, S.; Polok, K.; Fronczek, J.; Szczeklik, W.; Piticaru, J.; Mammen, M.J.; Alshamsi, F.; Eikelboom, J.; Belley-Cote, E.; et al. Venous and arterial thrombosis in COVID-19: An updated narrative review. J. Infect. Public Health 2022, 15, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; Munoz Martin, A.J. Thrombosis and Coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742. [Google Scholar] [CrossRef]
- Hadid, T.; Kafri, Z.; Al-Katib, A. Coagulation and anticoagulation in COVID-19. Blood Rev. 2021, 47, 100761. [Google Scholar] [CrossRef]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L. Thrombotic Regulation from the Endothelial Cell Perspectives. Arter. Thromb. Vasc. Biol. 2018, 38, e90–e95. [Google Scholar] [CrossRef] [PubMed]
- Sathler, P.C. Hemostatic abnormalities in COVID-19: A guided review. An. Acad. Bras. Cienc. 2020, 92, e20200834. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W. Endothelium—Role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Jalloh, S.; Olejnik, J.; Berrigan, J.; Nisa, A.; Suder, E.L.; Akiyama, H.; Lei, M.; Ramaswamy, S.; Tyagi, S.; Bushkin, Y.; et al. CD169-mediated restrictive SARS-CoV-2 infection of macrophages induces pro-inflammatory responses. PLoS Pathog. 2022, 18, e1010479. [Google Scholar] [CrossRef]
- Nascimento Conde, J.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 to Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 2020, 11, e03185-20. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Lei, T.; Patel, P.S.; Lee, C.H.; Monaghan-Nichols, P.; Xin, H.B.; Qiu, J.; Fu, M. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by Simvastatin. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xu, S.W.; Ilyas, I.; Weng, J.P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2022, 44, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Gue, Y.X.; Gorog, D.A. Reduction in ACE2 may mediate the prothrombotic phenotype in COVID-19. Eur. Heart J. 2020, 41, 3198–3199. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Cao, J.; Cai, J.P.; Wu, J.; Huang, J.; Asthana, P.; Wong, S.K.K.; Ye, Z.-W.; Gurung, S.; Zhang, Y.; et al. Control of SARS-CoV-2 infection by MT1-MMP-mediated shedding of ACE2. Nat. Commun. 2022, 13, 7907. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833. [Google Scholar] [CrossRef]
- Du, F.; Liu, B.; Zhang, S. COVID-19: The role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis 2021, 51, 313–329. [Google Scholar] [CrossRef]
- Inciardi, R.M.; Adamo, M.; Lupi, L.; Cani, D.S.; Di Pasquale, M.; Tomasoni, D.; Italia, L.; Zaccone, G.; Tedino, C.; Fabbricatore, D.; et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur. Heart J. 2020, 14, 1821–1829, Erratum in Eur. Heart J. 2020, 41, 4591. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Zamai, L. The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells 2020, 9, 1704. [Google Scholar] [CrossRef]
- Zamai, L. Upregulation of the Renin-Angiotensin System Pathways and SARS-CoV-2 Infection: The Rationale for the Administration of Zinc-Chelating Agents in COVID-19 Patients. Cells 2021, 10, 506. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Liu, Y.M.; Hu, J. Single-cell analysis of adult human heart across healthy and cardiovascular disease patients reveals the cellular landscape underlying SARS-CoV-2 invasion of myocardial tissue through ACE2. J. Transl. Med. 2023, 21, 358. [Google Scholar] [CrossRef]
- Patel, S.K.; Juno, J.A.; Lee, W.S.; Wragg, K.M.; Hogarth, P.M.; Kent, S.J.; Burrell, L.M. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: Implications for COVID-19 pathogenesis and consequences. Eur. Respir. J. 2021, 57, 2003730. [Google Scholar] [CrossRef]
- Benedetti, S.; Sisti, D.; Vandini, D.; Barocci, S.; Sudano, M.; Carlotti, E.; Teng, J.L.L.; Zamai, L. Circulating ACE2 level and zinc/albumin ratio as potential biomarkers for a precision medicine approach to COVID-19. Adv. Biol. Regul. 2023, 89, 100973. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lu, B.; Li, Y.; Yuan, S.; Zhuang, Z.; Li, G.; Wang, D.; Ma, L.; Zhu, J.; Zhao, J.; et al. P21-activated kinase 1 (PAK1)-mediated cytoskeleton rearrangement promotes SARS-CoV-2 entry and ACE2 autophagic degradation. Signal Transduct. Target. Ther. 2023, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; von Bruhl, M.L.; Barocke, V.; Cullen, P.; Mayer, K.; Okrojek, R.; Steinhart, A.; Ahmad, Z.; Kremmer, E.; Nieswandt, B.; et al. EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J. Thromb. Haemost. 2011, 9, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jin, R.; Zhu, X.; Yan, J.; Li, G. Function of CD147 in atherosclerosis and atherothrombosis. J. Cardiovasc. Transl. Res. 2015, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Scheuer, W.; Eggle, D.; Klostermann, S.; Stockinger, H. Cancer-related issues of CD147. Cancer Genom. Proteom. 2010, 7, 157–169. [Google Scholar]
- Khayati, F.; Perez-Cano, L.; Maouche, K.; Sadoux, A.; Boutalbi, Z.; Podgorniak, M.P.; Maskos, U.; Setterblad, N.; Janin, A.; Calvo, F.; et al. EMMPRIN/CD147 is a novel coreceptor of VEGFR-2 mediating its activation by VEGF. Oncotarget 2015, 6, 9766–9780. [Google Scholar] [CrossRef] [PubMed]
- Quemener, C.; Gabison, E.E.; Naimi, B.; Lescaille, G.; Bougatef, F.; Podgorniak, M.P.; Labarchède, G.; Lebbé, C.; Calvo, F.; Menashi, S.; et al. Extracellular matrix metalloproteinase inducer up-regulates the urokinase-type plasminogen activator system promoting tumor cell invasion. Cancer Res. 2007, 67, 9–15. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef]
- Bhattacharjee, G.; Ahamed, J.; Pedersen, B.; El-Sheikh, A.; Mackman, N.; Ruf, W.; Liu, C.; Edgington, T.S. Regulation of tissue factor--mediated initiation of the coagulation cascade by cell surface grp78. Arter. Thromb. Vasc. Biol. 2005, 25, 1737–1743. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef]
- Watson, L.M.; Chan, A.K.; Berry, L.R.; Li, J.; Sood, S.K.; Dickhout, J.G.; Xu, L.; Werstuck, G.H.; Bajzar, L.; Klamut, H.J.; et al. Overexpression of the 78-kDa glucose-regulated protein/immunoglobulin-binding protein (GRP78/BiP) inhibits tissue factor procoagulant activity. J. Biol. Chem. 2003, 278, 17438–17447. [Google Scholar] [CrossRef] [PubMed]
- Lenin, R.; Nagy, P.G.; Jha, K.A.; Gangaraju, R. GRP78 translocation to the cell surface and O-GlcNAcylation of VE-Cadherin contribute to ER stress-mediated endothelial permeability. Sci. Rep. 2019, 9, 10783. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.; Kolloli, A.; Kumar, R.; Husain, S.; Soteropoulos, P.; Chang, T.L.; Subbian, S. Comprehensive Analysis of Disease Pathology in Immunocompetent and Immunocompromised Hosts following Pulmonary SARS-CoV-2 Infection. Biomedicines 2022, 10, 1343. [Google Scholar] [CrossRef]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell 2020, 27, 962–973.e7. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Bailey, A.L.; Kim, A.S.; Chen, R.E.; Diamond, M.S. Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology 2020, 548, 39–48. [Google Scholar] [CrossRef]
- Subbian, S.; Bandyopadhyay, N.; Tsenova, L.; O’Brien, P.; Khetani, V.; Kushner, N.L.; Peixoto, B.; Soteropoulos, P.; Bader, J.S.; Karakousis, P.C.; et al. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun. Signal. 2013, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Miller, C.M.; Akiyama, H.; Agosto, L.M.; Emery, A.; Ettinger, C.R.; Swanstrom, R.I.; Henderson, A.J.; Gummuluru, S. Virion-Associated Vpr Alleviates a Postintegration Block to HIV-1 Infection of Dendritic Cells. J. Virol. 2017, 91, e00051-17. [Google Scholar] [CrossRef]
- Perera, R.; Ko, R.; Tsang, O.T.Y.; Hui, D.S.C.; Kwan, M.Y.M.; Brackman, C.J.; To, E.M.W.; Yen, H.-L.; Leung, K.; Cheng, S.M.S.; et al. Evaluation of a SARS-CoV-2 Surrogate Virus Neutralization Test for Detection of Antibody in Human, Canine, Cat, and Hamster Sera. J. Clin. Microbiol. 2021, 59. [Google Scholar] [CrossRef]
- Subbian, S.; Tsenova, L.; Holloway, J.; Peixoto, B.; O’Brien, P.; Dartois, V.; Khetani, V.; Zeldis, J.B.; Kaplan, G. Adjunctive Phosphodiesterase-4 Inhibitor Therapy Improves Antibiotic Response to Pulmonary Tuberculosis in a Rabbit Model. EBioMedicine 2016, 4, 104–114. [Google Scholar] [CrossRef]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.; Nam, J.Y.; Blancato, J.K. Fluorescence In Situ Hybridization of Cells, Chromosomes, and Formalin-Fixed Paraffin-Embedded Tissues. Methods Mol. Biol. 2017, 1606, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Traylor-Knowles, N. In Situ Hybridization Techniques for Paraffin-Embedded Adult Coral Samples. J. Vis. Exp. 2018, 138, e57853. [Google Scholar] [CrossRef]
- Hu, W.; Song, X.; Yu, H.; Zhao, L.; Zhao, Y.; Zhao, Y. Further comments on the role of ACE-2 positive macrophages in human lung. Cytom. Part A 2023, 103, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Bujan, J.; Gimeno, M.J.; Prieto, A.; Pascual, G.; Bellon, J.M.; Alvarez-Mon, M. Modulation of PECAM-1 (CD31) expression in human endothelial cells: Effect of IFNgamma and IL-10. J. Vasc. Res. 1999, 36, 106–113. [Google Scholar] [CrossRef]
- Shaw, S.K.; Perkins, B.N.; Lim, Y.C.; Liu, Y.; Nusrat, A.; Schnell, F.J.; Parkos, C.A.; Luscinskas, F.W. Reduced expression of junctional adhesion molecule and platelet/endothelial cell adhesion molecule-1 (CD31) at human vascular endothelial junctions by cytokines tumor necrosis factor-alpha plus interferon-gamma does not reduce leukocyte transmigration under flow. Am. J. Pathol. 2001, 159, 2281–2291. [Google Scholar] [CrossRef]
- Helal, M.A.; Shouman, S.; Abdelwaly, A.; Elmehrath, A.O.; Essawy, M.; Sayed, S.M.; Saleh, A.H.; El-Badri, N. Molecular basis of the potential interaction of SARS-CoV-2 spike protein to CD147 in COVID-19 associated-lymphopenia. J. Biomol. Struct. Dyn. 2022, 40, 1109–1119. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Semeraro, N.; Colucci, M. The Prothrombotic State Associated with SARS-CoV-2 Infection: Pathophysiological Aspects. Mediterr. J. Hematol. Infect. Dis. 2021, 13, e2021045. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Popova, P.I.; Avdonin, P.P.; Kudryavtsev, I.V.; Serebryakova, M.K.; Korf, E.A.; Avdonin, P.V. Markers of Endothelial Cells in Normal and Pathological Conditions. Biochem. Mosc. Suppl. Ser. A Membr. Cell Biol. 2020, 14, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.A.; Colley, L.; Agbaedeng, T.A.; Ellison-Hughes, G.M.; Ross, M.D. Vascular Manifestations of COVID-19—Thromboembolism and Microvascular Dysfunction. Front. Cardiovasc. Med. 2020, 7, 598400. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Osman, I.O.; Melenotte, C.; Brouqui, P.; Million, M.; Lagier, J.C.; Parola, P.; Stein, A.; La Scola, B.; Meddeb, L.; Mege, J.L.; et al. Expression of ACE2, Soluble ACE2, Angiotensin I, Angiotensin II and Angiotensin-(1-7) Is Modulated in COVID-19 Patients. Front. Immunol. 2021, 12, 625732. [Google Scholar] [CrossRef]
- Jover, E.; Matilla, L.; Garaikoetxea, M.; Fernandez-Celis, A.; Muntendam, P.; Jaisser, F.; Rossignol, P.; López-Andrés, N. Beneficial Effects of Mineralocorticoid Receptor Pathway Blockade against Endothelial Inflammation Induced by SARS-CoV-2 Spike Protein. Biomedicines 2021, 9, 639. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, J.; Wang, P.H.; Yang, N.; Huang, J.; Ou, J.; Xu, T.; Zhao, X.; Liu, T.; Huang, X.; et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166260. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells. Biomedicines 2021, 9, 1220. [Google Scholar] [CrossRef] [PubMed]
- Biering, S.B.; Gomes de Sousa, F.T.; Tjang, L.V.; Pahmeier, F.; Zhu, C.; Ruan, R.; Blanc, S.F.; Patel, T.S.; Worthington, C.M.; Glasner, D.R.; et al. SARS-CoV-2 Spike triggers barrier dysfunction and vascular leak via integrins and TGF-beta signaling. Nat. Commun. 2022, 13, 7630. [Google Scholar] [CrossRef] [PubMed]
- Colunga Biancatelli, R.M.L.; Solopov, P.A.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Kappa18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef]
- Zheng, X.; Li, W.; Gu, J.M.; Qu, D.; Ferrell, G.L.; Esmon, N.L.; Esmon, C.T. Effects of membrane and soluble EPCR on the hemostatic balance and endotoxemia in mice. Blood 2007, 109, 1003–1009. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Aguiar, J.A.; Tremblay, B.J.; Mansfield, M.J.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Cao, Q.; Dvorkin-Gheva, A.; et al. Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 2020, 56, 2001123. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, S.; Gou, J.; Wen, Y.; Fan, L.; Zhou, J.; Zhou, G.; Xu, G.; Zhang, Z. Spike-mediated ACE2 down-regulation was involved in the pathogenesis of SARS-CoV-2 infection. J. Infect. 2022, 85, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhu, Q.; Fox, D.M.; Gao, C.; Stanley, S.A.; Luo, K. SARS-CoV-2 down-regulates ACE2 through lysosomal degradation. Mol. Biol. Cell. 2022, 33, ar147. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Hsueh, C.H.; Hsu, P.M.; Huang, R.H.; Tsai, C.Y.; Chung, N.H.; Chow, Y.H.; Tan, T.H. SARS-CoV-2 spike protein enhances MAP4K3/GLK-induced ACE2 stability in COVID-19. EMBO Mol. Med. 2022, 14, e15904. [Google Scholar] [CrossRef] [PubMed]
- Gheware, A.; Ray, A.; Rana, D.; Bajpai, P.; Nambirajan, A.; Arulselvi, S.; Mathur, P.; Trikha, A.; Arava, S.; Das, P.; et al. ACE2 protein expression in lung tissues of severe COVID-19 infection. Sci. Rep. 2022, 12, 4058. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Canonico, B.; Nordi, E.; Vandini, D.; Barocci, S.; Benedetti, S.; Carlotti, E.; Zamai, L. Which ones, when and why should renin-angiotensin system inhibitors work against COVID-19? Adv. Biol. Regul. 2021, 81, 100820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Luo, W.; Huang, L.; Xiao, J.; Li, F.; Qin, S.; Song, X.; Wu, Y.; Zeng, Q.; et al. A comprehensive investigation of the mRNA and protein level of ACE2, the putative receptor of SARS-CoV-2, in human tissues and blood cells. Int. J. Med. Sci. 2020, 17, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Dielis, A.W.; Smid, M.; Spronk, H.M.; Hamulyak, K.; Kroon, A.A.; ten Cate, H.; de Leeuw, P.W. The prothrombotic paradox of hypertension: Role of the renin-angiotensin and kallikrein-kinin systems. Hypertension 2005, 46, 1236–1242. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nisa, A.; Kumar, R.; Ramasamy, S.; Kolloli, A.; Olejnik, J.; Jalloh, S.; Gummuluru, S.; Subbian, S.; Bushkin, Y. Modulations of Homeostatic ACE2, CD147, GRP78 Pathways Correlate with Vascular and Endothelial Performance Markers during Pulmonary SARS-CoV-2 Infection. Cells 2024, 13, 432. https://doi.org/10.3390/cells13050432
Nisa A, Kumar R, Ramasamy S, Kolloli A, Olejnik J, Jalloh S, Gummuluru S, Subbian S, Bushkin Y. Modulations of Homeostatic ACE2, CD147, GRP78 Pathways Correlate with Vascular and Endothelial Performance Markers during Pulmonary SARS-CoV-2 Infection. Cells. 2024; 13(5):432. https://doi.org/10.3390/cells13050432
Chicago/Turabian StyleNisa, Annuurun, Ranjeet Kumar, Santhamani Ramasamy, Afsal Kolloli, Judith Olejnik, Sallieu Jalloh, Suryaram Gummuluru, Selvakumar Subbian, and Yuri Bushkin. 2024. "Modulations of Homeostatic ACE2, CD147, GRP78 Pathways Correlate with Vascular and Endothelial Performance Markers during Pulmonary SARS-CoV-2 Infection" Cells 13, no. 5: 432. https://doi.org/10.3390/cells13050432
APA StyleNisa, A., Kumar, R., Ramasamy, S., Kolloli, A., Olejnik, J., Jalloh, S., Gummuluru, S., Subbian, S., & Bushkin, Y. (2024). Modulations of Homeostatic ACE2, CD147, GRP78 Pathways Correlate with Vascular and Endothelial Performance Markers during Pulmonary SARS-CoV-2 Infection. Cells, 13(5), 432. https://doi.org/10.3390/cells13050432