
Role of Inflammatory Mechanisms in Major Depressive Disorder: From Etiology to Potential Pharmacological Targets


 ,
,  and
and 
Abstract
1. Introduction
2. Neurobiology of Major Depressive Disorder
3. Neuroinflammation: Function of Glial Cells
3.1. Microglia
3.2. Astrocytes
3.3. Changes in the Blood–Brain Barrier Due to Glial Activation
4. Role of the Peripheral Immune System in MDD
5. Gut Microbiota
6. Anti-Inflammatory Properties of Antidepressants
7. Antidepressant Effects of Anti-Inflammatory Drugs
8. Alternative MDD Therapeutic Approaches with Anti-Inflammatory Properties
8.1. Physical Exercise
8.2. Agmatine
8.3. Probiotics
8.4. Vitamin C (Ascorbic Acid)
8.5. Vitamin D
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. DSM-5 Task Force. In Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- World Health Organization. Depressive Disorder (Depression). 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 12 January 2024).
- Marx, W.; Penninx, B.W.J.H.; Solmi, M.; Furukawa, T.A.; Firth, J.; Carvalho, A.F.; Berk, M. Major depressive disorder. Nat. Rev. Dis. Primers 2023, 9, 44. [Google Scholar] [CrossRef]
- Hamon, M.; Blier, P. Monoamine neurocircuitry in depression and strategies for new treatments. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 45, 54–63. [Google Scholar] [CrossRef]
- Caldiroli, A.; Capuzzi, E.; Tagliabue, I.; Capellazzi, M.; Marcatili, M.; Mucci, F.; Colmegna, F.; Clerici, M.; Buoli, M.; Dakanalis, A. Augmentative Pharmacological Strategies in Treatment-Resistant Major Depression: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 13070. [Google Scholar] [CrossRef]
- Hodes, G.E.; Kana, V.; Menard, C.; Merad, M.; Russo, S.J. Neuroimmune mechanisms of depression. Nat. Neurosci. 2015, 18, 1386–1393. [Google Scholar] [CrossRef]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Patil, C.R.; Suryakant Gawli, C.; Bhatt, S. Targeting inflammatory pathways for treatment of the major depressive disorder. Drug Discov. Today 2023, 28, 103697. [Google Scholar] [CrossRef] [PubMed]
- Fries, G.R.; Saldana, V.A.; Finnstein, J.; Rein, T. Molecular pathways of major depressive disorder converge on the synapse. Mol. Psychiatry 2023, 28, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Baxter, L.J.; Lewis, G.; Jones, P.B.; Khandaker, G.M. Prevalence of low-grade inflammation in depression: A systematic review and meta-analysis of CRP levels. Psychol. Med. 2019, 49, 1958–1970. [Google Scholar] [CrossRef]
- Smith, K.J.; Au, B.; Ollis, L.; Schmitz, N. The association between C-reactive protein, Interleukin-6 and depression among older adults in the community: A systematic review and meta-analysis. Exp. Gerontol. 2018, 102, 109–132. [Google Scholar] [CrossRef]
- Colasanto, M.; Madigan, S.; Korczak, D.J. Depression and inflammation among children and adolescents: A meta-analysis. J. Affect. Disord. 2020, 277, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.; Mondelli, V.; Pariante, C.M. Genetic Contributions of Inflammation to Depression. Neuropsychopharmacology 2017, 42, 81–98. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Ronconi, G.; Conti, C.M.; Kritas, S.K.; Mastrangelo, F.; Tettamanti, L.; Theoharides, T.C. Impact of mast cells in depression disorder: Inhibitory effect of IL-37 (new frontiers). Immunol. Res. 2018, 66, 323–331. [Google Scholar] [CrossRef]
- Myint, A.M.; Schwarz, M.J.; Steinbusch, H.W.; Leonard, B.E. Neuropsychiatric disorders related to interferon and interleukins treatment. Metab. Brain Dis. 2009, 24, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A.; Walld, R.; Bolton, J.M.; Sareen, J.; Patten, S.B.; Singer, A.; Lix, L.M.; Hitchon, C.A.; El-Gabalawy, R.; Katz, A.; et al. Psychiatric comorbidity increases mortality in immune-mediated inflammatory diseases. Gen. Hosp. Psychiatry 2018, 53, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, J.D.; Liu, L.; Poirier, S.E.; Schaefer, B.; Poolacherla, R.; Burhan, A.M.; Sabesan, P.; St Lawrence, K.; Théberge, J.; Hicks, J.W.; et al. The functional and structural associations of aberrant microglial activity in major depressive disorder. J. Psychiatry Neurosci. 2022, 47, 197–208. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Bhatt, S.; Mahesh, R.; Devadoss, T.; Jindal, A. Neuropharmacological evaluation of a novel 5-HT3 receptor antagonist (4-benzylpiperazin-1-yl)(3-methoxyquinoxalin-2-yl) methanone (6g) on lipopolysaccharide-induced anxiety models in mice. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 101–106. [Google Scholar] [CrossRef]
- Berk, M.; Woods, R.L.; Nelson, M.R.; Shah, R.C.; Reid, C.M.; Storey, E.; Fitzgerald, S.; Lockery, J.E.; Wolfe, R.; Mohebbi, M.; et al. Effect of Aspirin vs Placebo on the Prevention of Depression in Older People: A Randomized Clinical Trial. JAMA Psychiatry 2020, 77, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Vreeburg, S.A.; Hoogendijk, W.J.; van Pelt, J.; Derijk, R.H.; Verhagen, J.C.; van Dyck, R.; Smit, J.H.; Zitman, F.G.; Penninx, B.W. Major depressive disorder and hypothalamic-pituitary-adrenal axis activity: Results from a large cohort study. Arch. Gen. Psychiatry 2009, 66, 617–626. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Leistner, C.; Menke, A. Hypothalamic-pituitary-adrenal axis and stress. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 175, pp. 55–64. [Google Scholar] [CrossRef]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2017, 22, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Ahmed, I.; Moktadir, A.A.; Nahar, Z.; Islam, M.S.; Shahid, S.F.B.; Islam, S.N.; Hasnat, A. Elevated serum levels of malondialdehyde and cortisol are associated with major depressive disorder: A case-control study. SAGE Open Med. 2018, 6, 2050312118773953. [Google Scholar] [CrossRef] [PubMed]
- Kennis, M.; Gerritsen, L.; van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective biomarkers of major depressive disorder: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 321–338. [Google Scholar] [CrossRef]
- Cattaneo, A.; Ferrari, C.; Turner, L.; Mariani, N.; Enache, D.; Hastings, C.; Kose, M.; Lombardo, G.; McLaughlin, A.P.; Nettis, M.A.; et al. Whole-blood expression of inflammasome- and glucocorticoid-related mRNAs correctly separates treatment-resistant depressed patients from drug-free and responsive patients in the BIODEP study. Transl. Psychiatry 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Ma, N.; Yan, D.; Wang, Y.; Zhao, X.; Zhang, Y.; Zhang, C. Effects of corticosterone on the expression of mature brain-derived neurotrophic factor (mBDNF) and proBDNF in the hippocampal dentate gyrus. Behav. Brain Res. 2019, 365, 150–156. [Google Scholar] [CrossRef]
- Kvarta, M.D.; Bradbrook, K.E.; Dantrassy, H.M.; Bailey, A.M.; Thompson, S.M. Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J. Neurophysiol. 2015, 114, 1713–1724. [Google Scholar] [CrossRef]
- Malek, H.; Ebadzadeh, M.M.; Safabakhsh, R.; Razavi, A.; Zaringhalam, J. Dynamics of the HPA axis and inflammatory cytokines: Insights from mathematical modeling. Comput. Biol. Med. 2015, 67, 1–12. [Google Scholar] [CrossRef]
- You, Z.; Luo, C.; Zhang, W.; Chen, Y.; He, J.; Zhao, Q.; Zuo, R.; Wu, Y. Pro- and anti-inflammatory cytokines expression in rat’s brain and spleen exposed to chronic mild stress: Involvement in depression. Behav. Brain Res. 2011, 225, 135–141. [Google Scholar] [CrossRef]
- Song, A.Q.; Gao, B.; Fan, J.J.; Zhu, Y.J.; Zhou, J.; Wang, Y.L.; Xu, L.Z.; Wu, W.N. NLRP1 inflammasome contributes to chronic stress-induced depressive-like behaviors in mice. J. Neuroinflamm. 2020, 17, 178. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, F.; Li, P.; Song, C. Low-Dose IL-2 Attenuated Depression-like Behaviors and Pathological Changes through Restoring the Balances between IL-6 and TGF-β and between Th17 and Treg in a Chronic Stress-Induced Mouse Model of Depression. Int. J. Mol. Sci. 2022, 23, 13856. [Google Scholar] [CrossRef]
- Anderson, G.; Kubera, M.; Duda, W.; Lasoń, W.; Berk, M.; Maes, M. Increased IL-6 trans-signaling in depression: Focus on the tryptophan catabolite pathway, melatonin and neuroprogression. Pharmacol. Rep. 2013, 65, 1647–1654. [Google Scholar] [CrossRef]
- Halaris, A.; Cook, J. The Glutamatergic System in Treatment-Resistant Depression and Comparative Effectiveness of Ketamine and Esketamine: Role of Inflammation? Adv. Exp. Med. Biol. 2023, 1411, 487–512. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Duman, R.S.; Li, N.; Liu, R.J.; Duric, V.; Aghajanian, G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012, 62, 35–41. [Google Scholar] [CrossRef]
- Réus, G.Z.; Nacif, M.P.; Abelaira, H.M.; Tomaz, D.B.; dos Santos, M.A.; Carlessi, A.S.; da Luz, J.R.; Gonçalves, R.C.; Vuolo, F.; Dal-Pizzol, F.; et al. Ketamine ameliorates depressive-like behaviors and immune alterations in adult rats following maternal deprivation. Neurosci. Lett. 2015, 584, 83–87. [Google Scholar] [CrossRef]
- Réus, G.Z.; Silva, R.H.; de Moura, A.B.; Presa, J.F.; Abelaira, H.M.; Abatti, M.; Vieira, A.; Pescador, B.; Michels, M.; Ignácio, Z.M.; et al. Early Maternal Deprivation Induces Microglial Activation, Alters Glial Fibrillary Acidic Protein Immunoreactivity and Indoleamine 2,3-Dioxygenase during the Development of Offspring Rats. Mol. Neurobiol. 2019, 56, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Kunugi, H. Gut Microbiota and Pathophysiology of Depressive Disorder. Ann. Nutr. Metab. 2021, 77 (Suppl. S2), 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Kanoujia, J.; Mohana Lakshmi, S.; Patil, C.R.; Gupta, G.; Chellappan, D.K.; Dua, K. Role of Brain-Gut-Microbiota Axis in Depression: Emerging Therapeutic Avenues. CNS Neurol. Disord. Drug Targets 2023, 22, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Gao, X.R.; Peng, L.; Ge, J.F. Crosstalk between the microbiota-gut-brain axis and depression. Heliyon 2020, 6, e04097. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Blacher, E.; Elinav, E.; Pettersson, S. Our Gut Microbiome: The Evolving Inner Self. Cell 2017, 171, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.T.; Wang, S.Q.; Su, J.; Xu, L.X.; Ji, Z.Y.; Zhang, R.Y.; Zhao, Q.W.; Ma, Z.Q.; Deng, X.Y.; Ma, S.P. Baicalin ameliorates neuroinflammation-induced depressive-like behavior through inhibition of toll-like receptor 4 expression via the PI3K/AKT/FoxO1 pathway. J. Neuroinflamm. 2019, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Réus, G.Z.; Jansen, K.; Titus, S.; Carvalho, A.F.; Gabbay, V.; Quevedo, J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: Evidences from animal and human studies. J. Psychiatr. Res. 2015, 68, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Sipahi, H.; Mat, A.F.; Ozhan, Y.; Aydin, A. The Interrelation between Oxidative Stress, Depression and Inflammation through the Kynurenine Pathway. Curr. Top. Med. Chem. 2023, 23, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Bao, C.H.; Wu, Y.; Liang, S.H.; Wang, D.; Wu, L.Y.; Huang, Y.; Liu, H.R.; Wu, H.G. Tryptophan-kynurenine metabolism: A link between the gut and brain for depression in inflammatory bowel disease. J. Neuroinflammation 2021, 18, 135. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Barone, P. The ‘Yin’ and the ‘Yang’ of the kynurenine pathway: Excitotoxicity and neuroprotection imbalance in stress-induced disorders. Behav. Pharmacol. 2019, 30, 163–186. [Google Scholar] [CrossRef]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in health and disease. Adv. Clin. Chem. 2020, 95, 165–218. [Google Scholar] [CrossRef]
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef]
- Hochstrasser, T.; Ullrich, C.; Sperner-Unterweger, B.; Humpel, C. Inflammatory stimuli reduce survival of serotonergic neurons and induce neuronal expression of indoleamine 2,3-dioxygenase in rat dorsal raphe nucleus organotypic brain slices. Neuroscience 2011, 184, 128–138. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112 Pt B, 237–247. [Google Scholar] [CrossRef]
- Gosselin, R.D.; Gibney, S.; O’Malley, D.; Dinan, T.G.; Cryan, J.F. Region specific decrease in glial fibrillary acidic protein immunoreactivity in the brain of a rat model of depression. Neuroscience 2009, 159, 915–925. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, L.A.; Mechawar, N. Implication of cerebral astrocytes in major depression: A review of fine neuroanatomical evidence in humans. Glia 2021, 69, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Zadori, D.; Toldi, J.; Fulop, F.; Klivenyi, P.; Vecsei, L. Manipulating kynurenic acid levels in the brain—On the edge between neuroprotection and cognitive dysfunction. Curr. Top. Med. Chem. 2012, 12, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Fujii, T.; Koga, N.; Hori, H.; Teraishi, T.; Hattori, K.; Noda, T.; Higuchi, T.; Motohashi, N.; Kunugi, H. Plasma L-tryptophan concentration in major depressive disorder: New data and meta-analysis. J. Clin. Psychiatry 2014, 75, e906–e915. [Google Scholar] [CrossRef] [PubMed]
- Wurfel, B.E.; Drevets, W.C.; Bliss, S.A.; McMillin, J.R.; Suzuki, H.; Ford, B.N.; Morris, H.M.; Teague, T.K.; Dantzer, R.; Savitz, J.B. Serum kynurenic acid is reduced in affective psychosis. Transl. Psychiatry 2017, 7, e1115. [Google Scholar] [CrossRef]
- Messaoud, A.; Mensi, R.; Douki, W.; Neffati, F.; Najjar, M.F.; Gobbi, G.; Valtorta, F.; Gaha, L.; Comai, S. Reduced peripheral availability of tryptophan and increased activation of the kynurenine pathway and cortisol correlate with major depression and suicide. World J. Biol. Psychiatry 2019, 20, 703–711. [Google Scholar] [CrossRef]
- Kouba, B.R.; Gil-Mohapel, J.; S Rodrigues, A.L. NLRP3 Inflammasome: From Pathophysiology to Therapeutic Target in Major Depressive Disorder. Int. J. Mol. Sci. 2022, 24, 133. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a microglial disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, A.S.; Borba, L.A.; Zugno, A.I.; Quevedo, J.; Réus, G.Z. Gut microbiota-brain axis in depression: The role of neuroinflammation. Eur. J. Neurosci. 2021, 53, 222–235. [Google Scholar] [CrossRef]
- Chagas, L.D.S.; Sandre, P.C.; Ribeiro E Ribeiro, N.C.A.; Marcondes, H.; Oliveira Silva, P.; Savino, W.; Serfaty, C.A. Environmental Signals on Microglial Function during Brain Development, Neuroplasticity, and Disease. Int. J. Mol. Sci. 2020, 21, 2111. [Google Scholar] [CrossRef]
- Wes, P.D.; Holtman, I.R.; Boddeke, E.W.; Möller, T.; Eggen, B.J. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016, 64, 197–213. [Google Scholar] [CrossRef]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Lemaitre, P.; Tareen, S.H.; Pasciuto, E.; Mascali, L.; Martirosyan, A.; Callaerts-Vegh, Z.; Poovathingal, S.; Dooley, J.; Holt, M.G.; Yshii, L.; et al. Molecular and cognitive signatures of ageing partially restored through synthetic delivery of IL2 to the brain. EMBO Mol. Med. 2023, 15, e16805. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573–2589.e2579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; He, H.; Qiao, Y.; Zhou, T.; Yi, S.; Zhang, L.; Mo, L.; Li, Y.; Jiang, W.; You, Z. Priming of microglia with IFN-γ impairs adult hippocampal neurogenesis and leads to depression-like behaviors and cognitive defects. Glia 2020, 68, 2674–2692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yan, M.; Pan, R.; Wang, Z.; Tao, X.; Li, C.; Xia, T.; Liu, X.; Chang, Q. Radix Polygalae extract exerts antidepressant effects in behavioral despair mice and chronic restraint stress-induced rats probably by promoting autophagy and inhibiting neuroinflammation. J. Ethnopharmacol. 2021, 265, 113317. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wu, G.; Wang, L.; Tan, Y.; Song, Z. Lactobacillus delbrueckii alleviates depression-like behavior through inhibiting toll-like receptor 4 (TLR4) signaling in mice. Ann. Transl. Med. 2021, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Y.; Yu, H.; Tian, Y.; Chen, X.; Chen, C.; Ren, Y.; Chen, Z.; Gong, X.; Cheng, K.; et al. Protective effect of Nr4a2 (Nurr1) against LPS-induced depressive-like behaviors via regulating activity of microglia and CamkII neurons in anterior cingulate cortex. Pharmacol. Res. 2023, 191, 106717. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiang, Y.; Zhu, Z.; Wang, W.; Jiang, Z.; Zhao, M.; Cheng, S.; Pan, F.; Liu, D.; Ho, R.C.M.; et al. Rifaximin-mediated gut microbiota regulation modulates the function of microglia and protects against CUMS-induced depression-like behaviors in adolescent rat. J. Neuroinflamm. 2021, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, Y.; Sun, Z.; Ren, S.; Liu, M.; Wang, G.; Yang, J. Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 2022, 19, 132. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.M.; Zanotti-Fregonara, P.; Fujita, M.; Newman, L.; Farmer, C.; Ballard, E.D.; Machado-Vieira, R.; Yuan, P.; Niciu, M.J.; Lyoo, C.H.; et al. PET radioligand binding to translocator protein (TSPO) is increased in unmedicated depressed subjects. EJNMMI Res. 2018, 8, 57. [Google Scholar] [CrossRef]
- Leulier, F.; Lemaitre, B. Toll-like receptors—Taking an evolutionary approach. Nat. Rev. Genet. 2008, 9, 165–178. [Google Scholar] [CrossRef]
- Rudzki, L.; Maes, M. The Microbiota-Gut-Immune-Glia (MGIG) Axis in Major Depression. Mol. Neurobiol. 2020, 57, 4269–4295. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016, 233, 1637–1650. [Google Scholar] [CrossRef] [PubMed]
- Herman, F.J.; Pasinetti, G.M. Principles of inflammasome priming and inhibition: Implications for psychiatric disorders. Brain Behav. Immun. 2018, 73, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-κB, IκB, and IKK: Integral Components of Immune System Signaling. Adv. Exp. Med. Biol. 2019, 1172, 207–226. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Montezuma, K.; Biojone, C.; Lisboa, S.F.; Cunha, F.Q.; Guimarães, F.S.; Joca, S.R. Inhibition of iNOS induces antidepressant-like effects in mice: Pharmacological and genetic evidence. Neuropharmacology 2012, 62, 485–491. [Google Scholar] [CrossRef]
- Lyu, D.; Wang, F.; Zhang, M.; Yang, W.; Huang, H.; Huang, Q.; Wu, C.; Qian, N.; Wang, M.; Zhang, H.; et al. Ketamine induces rapid antidepressant effects via the autophagy-NLRP3 inflammasome pathway. Psychopharmacology 2022, 239, 3201–3212. [Google Scholar] [CrossRef]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 1999, 424, 37–49. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, S.J.; Han, C.; Patkar, A.A.; Masand, P.S.; Pae, C.U. Oxidative/nitrosative stress and antidepressants: Targets for novel antidepressants. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 224–235. [Google Scholar] [CrossRef]
- Zhu, L.J.; Shi, H.J.; Chang, L.; Zhang, C.C.; Si, M.; Li, N.; Zhu, D.Y. nNOS-CAPON blockers produce anxiolytic effects by promoting synaptogenesis in chronic stress-induced animal models of anxiety. Br. J. Pharmacol. 2020, 177, 3674–3690. [Google Scholar] [CrossRef]
- Shi, H.J.; Wu, D.L.; Chen, R.; Li, N.; Zhu, L.J. Requirement of hippocampal DG nNOS-CAPON dissociation for the anxiolytic and antidepressant effects of fluoxetine. Theranostics 2022, 12, 3656–3675. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Yue, N.; Huang, H.; Zhu, X.; Han, Q.; Wang, Y.; Li, B.; Liu, Q.; Wu, G.; Zhang, Y.; Yu, J. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J. Neuroinflamm. 2017, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ha, H.; Jang, J.; Byun, Y. Recent Advances in the Development of Antidepressants Targeting the Purinergic P2X7 Receptor. Curr. Med. Chem. 2023, 30, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Chamera, K.; Trojan, E.; Szuster-Głuszczak, M.; Basta-Kaim, A. The Potential Role of Dysfunctions in Neuron-Microglia Communication in the Pathogenesis of Brain Disorders. Curr. Neuropharmacol. 2020, 18, 408–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, X.M.; Luo, Q.Q.; Huang, S.; Yang, Q.W.; Wang, F.X.; Ke, Y.; Qian, Z.M. CX3CL1/CX3CR1-mediated microglia activation plays a detrimental role in ischemic mice brain via p38MAPK/PKC pathway. J. Cereb. Blood Flow Metab. 2015, 35, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Zanier, E.R.; Marchesi, F.; Ortolano, F.; Perego, C.; Arabian, M.; Zoerle, T.; Sammali, E.; Pischiutta, F.; De Simoni, M.G. Fractalkine Receptor Deficiency Is Associated with Early Protection but Late Worsening of Outcome following Brain Trauma in Mice. J. Neurotrauma 2016, 33, 1060–1072. [Google Scholar] [CrossRef]
- Tang, M.M.; Lin, W.J.; Pan, Y.Q.; Li, Y.C. Fibroblast Growth Factor 2 Modulates Hippocampal Microglia Activation in a Neuroinflammation Induced Model of Depression. Front. Cell. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cui, Q.Q.; Hu, X.H.; Ye, J.; Liu, Z.C.; Mei, Y.X.; Wang, F.; Hu, Z.L.; Chen, J.G. CD200 in dentate gyrus improves depressive-like behaviors of mice through enhancing hippocampal neurogenesis via alleviation of microglia hyperactivation. J. Neuroinflamm. 2023, 20, 157. [Google Scholar] [CrossRef] [PubMed]
- Han, R.T.; Kim, R.D.; Molofsky, A.V.; Liddelow, S.A. Astrocyte-immune cell interactions in physiology and pathology. Immunity 2021, 54, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Wang, Y.; He, Y.; Wang, T.; Huang, X.H.; Zhao, C.M.; Zhang, L.; Li, S.W.; Wang, C.; Qu, Y.N.; et al. A1 astrocytes contribute to murine depression-like behavior and cognitive dysfunction, which can be alleviated by IL-10 or fluorocitrate treatment. J. Neuroinflamm. 2020, 17, 200. [Google Scholar] [CrossRef]
- Du Preez, A.; Onorato, D.; Eiben, I.; Musaelyan, K.; Egeland, M.; Zunszain, P.A.; Fernandes, C.; Thuret, S.; Pariante, C.M. Chronic stress followed by social isolation promotes depressive-like behaviour, alters microglial and astrocyte biology and reduces hippocampal neurogenesis in male mice. Brain Behav. Immun. 2021, 91, 24–47. [Google Scholar] [CrossRef]
- Novakovic, M.M.; Korshunov, K.S.; Grant, R.A.; Martin, M.E.; Valencia, H.A.; Budinger, G.R.S.; Radulovic, J.; Prakriya, M. Astrocyte reactivity and inflammation-induced depression-like behaviors are regulated by Orai1 calcium channels. Nat. Commun. 2023, 14, 5500. [Google Scholar] [CrossRef]
- Xu, S.X.; Xie, X.H.; Yao, L.; Wang, W.; Zhang, H.; Chen, M.M.; Sun, S.; Nie, Z.W.; Nagy, C.; Liu, Z. Human in vivo evidence of reduced astrocyte activation and neuroinflammation in patients with treatment-resistant depression following electroconvulsive therapy. Psychiatry Clin. Neurosci. 2023, 77, 653–664. [Google Scholar] [CrossRef]
- Hasel, P.; Rose, I.V.L.; Sadick, J.S.; Kim, R.D.; Liddelow, S.A. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 2021, 24, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fang, Y.; Zhang, Y.; Song, M.; Zhang, X.; Ding, X.; Yao, H.; Chen, M.; Sun, Y.; Ding, J.; et al. Microglial NLRP3 inflammasome activates neurotoxic astrocytes in depression-like mice. Cell Rep. 2022, 41, 111532. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Calì, C.; Bezzi, P. CXCR4-mediated glutamate exocytosis from astrocytes. J. Neuroimmunol. 2010, 224, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S. Role of glutamate receptors and glial cells in the pathophysiology of treatment-resistant depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 70, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ning, Y.; Hong, W.; Wang, J.; Liu, Z.; Li, M.D. Crosstalk Between Inflammation and Glutamate System in Depression: Signaling Pathway and Molecular Biomarkers for Ketamine’s Antidepressant Effect. Mol. Neurobiol. 2019, 56, 3484–3500. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Berliocchi, L.; Bano, D.; Nicotera, P. Ca2+ signals and death programmes in neurons. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2255–2258. [Google Scholar] [CrossRef]
- Hamano, H.; Nabekura, J.; Nishikawa, M.; Ogawa, T. Docosahexaenoic acid reduces GABA response in substantia nigra neuron of rat. J. Neurophysiol. 1996, 75, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Ineichen, B.V.; Okar, S.V.; Proulx, S.T.; Engelhardt, B.; Lassmann, H.; Reich, D.S. Perivascular spaces and their role in neuroinflammation. Neuron 2022, 110, 3566–3581. [Google Scholar] [CrossRef]
- Morris, G.; Fernandes, B.S.; Puri, B.K.; Walker, A.J.; Carvalho, A.F.; Berk, M. Leaky brain in neurological and psychiatric disorders: Drivers and consequences. Aust. N. Z. J. Psychiatry 2018, 52, 924–948. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Oxidative and Nitrosative Stress and Immune-Inflammatory Pathways in Patients with Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS). Curr. Neuropharmacol. 2014, 12, 168–185. [Google Scholar] [CrossRef]
- Vezzani, A.; Ravizza, T.; Balosso, S.; Aronica, E. Glia as a source of cytokines: Implications for neuronal excitability and survival. Epilepsia 2008, 49 (Suppl. S2), 24–32. [Google Scholar] [CrossRef]
- Fabene, P.F.; Navarro Mora, G.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar] [CrossRef]
- Archambault, A.S.; Sim, J.; Gimenez, M.A.; Russell, J.H. Defining antigen-dependent stages of T cell migration from the blood to the central nervous system parenchyma. Eur. J. Immunol. 2005, 35, 1076–1085. [Google Scholar] [CrossRef]
- Bielecki, B.; Mazurek, A.; Wolinski, P.; Glabinski, A. Expression of chemokine receptors CCR7 and CCR8 in the CNS during ChREAE. Scand. J. Immunol. 2007, 66, 383–392. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Sagar, D.; Lamontagne, A.; Foss, C.A.; Khan, Z.K.; Pomper, M.G.; Jain, P. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood-brain barrier through paracellular transmigration and ERK activation. J. Neuroinflamm. 2012, 9, 245. [Google Scholar] [CrossRef]
- Mills Ko, E.; Ma, J.H.; Guo, F.; Miers, L.; Lee, E.; Bannerman, P.; Burns, T.; Ko, D.; Sohn, J.; Soulika, A.M.; et al. Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J. Neuroinflamm. 2014, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rodriguez, E.M.; Beurel, E. Blood brain barrier and inflammation in depression. Neurobiol. Dis. 2022, 175, 105926. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Blood-brain barrier associated tight junction disruption is a hallmark feature of major psychiatric disorders. Transl. Psychiatry 2020, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Geraets, A.F.J.; van Agtmaal, M.J.M.; Stehouwer, C.D.A.; Sörensen, B.M.; Berendschot, T.T.J.M.; Webers, C.A.B.; Schaper, N.C.; Henry, R.M.A.; van der Kallen, C.J.H.; Eussen, S.J.P.M.; et al. Association of Markers of Microvascular Dysfunction with Prevalent and Incident Depressive Symptoms: The Maastricht Study. Hypertension 2020, 76, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Nettis, M.A.; Pariante, C.M. Is there neuroinflammation in depression? Understanding the link between the brain and the peripheral immune system in depression. Int. Rev. Neurobiol. 2020, 152, 23–40. [Google Scholar] [CrossRef]
- Sakamoto, S.; Zhu, X.; Hasegawa, Y.; Karma, S.; Obayashi, M.; Alway, E.; Kamiya, A. Inflamed brain: Targeting immune changes and inflammation for treatment of depression. Psychiatry Clin. Neurosci. 2021, 75, 304–311. [Google Scholar] [CrossRef]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef]
- Savitz, J.; Harrison, N.A. Interoception and Inflammation in Psychiatric Disorders. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 514–524. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Hepgul, N.; Pariante, C.M. Inflammation and depression. Curr. Top. Behav. Neurosci. 2013, 14, 135–151. [Google Scholar] [CrossRef]
- Kaster, M.P.; Gadotti, V.M.; Calixto, J.B.; Santos, A.R.; Rodrigues, A.L. Depressive-like behavior induced by tumor necrosis factor-α in mice. Neuropharmacology 2012, 62, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.; Dalmagro, A.P.; Wolin, I.A.V.; Kaster, M.P.; Rodrigues, A.L.S. The resilient phenotype elicited by ketamine against inflammatory stressors-induced depressive-like behavior is associated with NLRP3-driven signaling pathway. J. Psychiatr. Res. 2021, 144, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Smaniotto, T.; Casaril, A.M.; de Andrade Lourenço, D.; Sousa, F.S.; Seixas, F.K.; Collares, T.; Woloski, R.; da Silva Pinto, L.; Alves, D.; Savegnago, L. Intranasal administration of interleukin-4 ameliorates depression-like behavior and biochemical alterations in mouse submitted to the chronic unpredictable mild stress: Modulation of neuroinflammation and oxidative stress. Psychopharmacology 2023, 240, 935–950. [Google Scholar] [CrossRef]
- Anacker, C.; Zunszain, P.A.; Carvalho, L.A.; Pariante, C.M. The glucocorticoid receptor: Pivot of depression and of antidepressant treatment? Psychoneuroendocrinology 2011, 36, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R.I.; Cogswell, P.C.; Lofquist, A.K.; Baldwin, A.S. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 1995, 270, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Widén, C.; Gustafsson, J.A.; Wikström, A.C. Cytosolic glucocorticoid receptor interaction with nuclear factor-kappa B proteins in rat liver cells. Biochem. J. 2003, 373 Pt 1, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Dragoş, D.; Tănăsescu, M.D. The effect of stress on the defense systems. J. Med. Life 2010, 3, 10–18. [Google Scholar]
- Knezevic, E.; Nenic, K.; Milanovic, V.; Knezevic, N.N. The Role of Cortisol in Chronic Stress, Neurodegenerative Diseases, and Psychological Disorders. Cells 2023, 12, 2726. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Luborsky, L.; McKay, J.R.; Rosenthal, R.; Houldin, A.; Tax, A.; McCorkle, R.; Seligman, D.A.; Schmidt, K. The relationship of depression and stressors to immunological assays: A meta-analytic review. Brain Behav. Immun. 2001, 15, 199–226. [Google Scholar] [CrossRef]
- Alvarez-Mon, M.A.; Gómez-Lahoz, A.M.; Orozco, A.; Lahera, G.; Diaz, D.; Ortega, M.A.; Albillos, A.; Quintero, J.; Aubá, E.; Monserrat, J.; et al. Expansion of CD4 T Lymphocytes Expressing Interleukin 17 and Tumor Necrosis Factor in Patients with Major Depressive Disorder. J. Pers. Med. 2021, 11, 220. [Google Scholar] [CrossRef]
- Beurel, E.; Harrington, L.E.; Jope, R.S. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol. Psychiatry 2013, 73, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Suh, Y.H.; Chang, K.A. Interleukin-17 induced by cumulative mild stress promoted depression-like behaviors in young adult mice. Mol. Brain 2021, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wei, B.; Li, L.; Wang, B.; Sun, M. Th17 cells and inflammation in neurological disorders: Possible mechanisms of action. Front. Immunol. 2022, 13, 932152. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mao, M.; Zhu, L.; Sun, Q.; Tong, J.; Zhou, Z. IL-17A drives cognitive aging probably via inducing neuroinflammation and theta oscillation disruption in the hippocampus. Int. Immunopharmacol. 2022, 108, 108898. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, B.; Qiu, W.; Yang, L.; Hu, B.; Tian, X.; Yang, H. Altered expression of CD4(+)CD25(+) regulatory T cells and its 5-HT(1a) receptor in patients with major depression disorder. J. Affect. Disord. 2010, 124, 68–75. [Google Scholar] [CrossRef]
- Freier, E.; Weber, C.S.; Nowottne, U.; Horn, C.; Bartels, K.; Meyer, S.; Hildebrandt, Y.; Luetkens, T.; Cao, Y.; Pabst, C.; et al. Decrease of CD4(+)FOXP3(+) T regulatory cells in the peripheral blood of human subjects undergoing a mental stressor. Psychoneuroendocrinology 2010, 35, 663–673. [Google Scholar] [CrossRef]
- Gao, X.; Tang, Y.; Kong, L.; Fan, Y.; Wang, C.; Wang, R. Treg cell: Critical role of regulatory T-cells in depression. Pharmacol. Res. 2023, 195, 106893. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, R.; Li, X.; Sun, W.; Qu, M.; Tang, Q.; Yang, X.; Zhang, S. Shen-Qi-Jie-Yu-Fang exerts effects on a rat model of postpartum depression by regulating inflammatory cytokines and CD4(+)CD25(+) regulatory T cells. Neuropsychiatr. Dis. Treat. 2016, 12, 883–896. [Google Scholar] [CrossRef]
- Lee, J.E.; Lee, J.M.; Park, Y.J.; Kim, B.S.; Jeon, Y.T.; Chung, Y. Inhibition of autoimmune Th17 cell responses by pain killer ketamine. Oncotarget 2017, 8, 89475–89485. [Google Scholar] [CrossRef][Green Version]
- Schneider-Schaulies, J.; Beyersdorf, N. CD4+ Foxp3+ regulatory T cell-mediated immunomodulation by anti-depressants inhibiting acid sphingomyelinase. Biol. Chem. 2018, 399, 1175–1182. [Google Scholar] [CrossRef]
- Benros, M.E.; Waltoft, B.L.; Nordentoft, M.; Ostergaard, S.D.; Eaton, W.W.; Krogh, J.; Mortensen, P.B. Autoimmune diseases and severe infections as risk factors for mood disorders: A nationwide study. JAMA Psychiatry 2013, 70, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Pryce, C.R.; Fontana, A. Depression in Autoimmune Diseases. Curr. Top. Behav. Neurosci. 2017, 31, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Laske, C.; Zank, M.; Klein, R.; Stransky, E.; Batra, A.; Buchkremer, G.; Schott, K. Autoantibody reactivity in serum of patients with major depression, schizophrenia and healthy controls. Psychiatry Res. 2008, 158, 83–86. [Google Scholar] [CrossRef]
- Andersson, N.W.; Gustafsson, L.N.; Okkels, N.; Taha, F.; Cole, S.W.; Munk-Jørgensen, P.; Goodwin, R.D. Depression and the risk of autoimmune disease: A nationally representative, prospective longitudinal study. Psychol. Med. 2015, 45, 3559–3569. [Google Scholar] [CrossRef]
- Bialek, K.; Czarny, P.; Strycharz, J.; Sliwinski, T. Major depressive disorders accompanying autoimmune diseases—Response to treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 95, 109678. [Google Scholar] [CrossRef]
- Leighton, S.P.; Nerurkar, L.; Krishnadas, R.; Johnman, C.; Graham, G.J.; Cavanagh, J. Chemokines in depression in health and in inflammatory illness: A systematic review and meta-analysis. Mol. Psychiatry 2018, 23, 48–58. [Google Scholar] [CrossRef]
- Brenhouse, H.C.; Schwarz, J.M. Immunoadolescence: Neuroimmune development and adolescent behavior. Neurosci. Biobehav. Rev. 2016, 70, 288–299. [Google Scholar] [CrossRef]
- Toenders, Y.J.; Laskaris, L.; Davey, C.G.; Berk, M.; Milaneschi, Y.; Lamers, F.; Penninx, B.W.J.H.; Schmaal, L. Inflammation and depression in young people: A systematic review and proposed inflammatory pathways. Mol. Psychiatry 2022, 27, 315–327. [Google Scholar] [CrossRef]
- D’Acunto, G.; Nageye, F.; Zhang, J.; Masi, G.; Cortese, S. Inflammatory Cytokines in Children and Adolescents with Depressive Disorders: A Systematic Review and Meta-Analysis. J. Child Adolesc. Psychopharmacol. 2019, 29, 362–369. [Google Scholar] [CrossRef]
- Rainville, J.R.; Hodes, G.E. Inflaming sex differences in mood disorders. Neuropsychopharmacology 2019, 44, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, G.; Mondelli, V.; Dazzan, P.; Pariante, C.M. Sex hormones and immune system: A possible interplay in affective disorders? A systematic review. J. Affect. Disord. 2021, 290, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Birur, B.; Amrock, E.M.; Shelton, R.C.; Li, L. Sex Differences in the Peripheral Immune System in Patients with Depression. Front. Psychiatry 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, P.; Sagar, R.; Mehta, M.; Sharma, S.; Subramanium, A.; Shamshi, F.; Sengupta, U.; Pandey, R.M.; Mukhopadhyay, A.K. Serum cytokines and anxiety in adolescent depression patients: Gender effect. Psychiatry Res. 2015, 229, 374–380. [Google Scholar] [CrossRef]
- Köhler-Forsberg, O.; Buttenschøn, H.N.; Tansey, K.E.; Maier, W.; Hauser, J.; Dernovsek, M.Z.; Henigsberg, N.; Souery, D.; Farmer, A.; Rietschel, M.; et al. Association between C-reactive protein (CRP) with depression symptom severity and specific depressive symptoms in major depression. Brain Behav. Immun. 2017, 62, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Moriarity, D.P.; Giollabhui, N.M.; Ellman, L.M.; Klugman, J.; Coe, C.L.; Abramson, L.Y.; Alloy, L.B. Inflammatory Proteins Predict Change in Depressive Symptoms in Male and Female Adolescents. Clin. Psychol. Sci. 2019, 7, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.H.; Chang, K.A. Sex Difference in Peripheral Inflammatory Biomarkers in Drug-Naïve Patients with Major Depression in Young Adulthood. Biomedicines 2021, 9, 708. [Google Scholar] [CrossRef] [PubMed]
- Debnath, M.; Berk, M.; Maes, M. Translational evidence for the Inflammatory Response System (IRS)/Compensatory Immune Response System (CIRS) and neuroprogression theory of major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110343. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Carvalho, A.F. The Compensatory Immune-Regulatory Reflex System (CIRS) in Depression and Bipolar Disorder. Mol. Neurobiol. 2018, 55, 8885–8903. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Meltzer, H.Y.; Bosmans, E.; Bergmans, R.; Vandoolaeghe, E.; Ranjan, R.; Desnyder, R. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J. Affect. Disord. 1995, 34, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Bosmans, E.; De Jongh, R.; Kenis, G.; Vandoolaeghe, E.; Neels, H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 1997, 9, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Feng, R.; Yang, Y. Changes in the serum levels of inflammatory cytokines in antidepressant drug-naïve patients with major depression. PLoS ONE 2018, 13, e0197267. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sánchez, G.; Becerril-Villanueva, E.; Arreola, R.; Martínez-Levy, G.; Hernández-Gutiérrez, M.E.; Velasco-Velásquez, M.A.; Alvarez-Herrera, S.; Cruz-Fuentes, C.; Palacios, L.; de la Peña, F.; et al. Inflammatory Profiles in Depressed Adolescents Treated with Fluoxetine: An 8-Week Follow-Up Open Study. Mediat. Inflamm. 2018, 2018, 4074051. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Ferencova, N.; Visnovcova, Z.; Ondrejka, I.; Funakova, D.; Hrtanek, I.; Kelcikova, S.; Tonhajzerova, I. Evaluation of Inflammatory Response System (IRS) and Compensatory Immune Response System (CIRS) in Adolescent Major Depression. J. Inflamm. Res. 2022, 15, 5959–5976. [Google Scholar] [CrossRef]
- Sowa-Kućma, M.; Styczeń, K.; Siwek, M.; Misztak, P.; Nowak, R.J.; Dudek, D.; Rybakowski, J.K.; Nowak, G.; Maes, M. Are there differences in lipid peroxidation and immune biomarkers between major depression and bipolar disorder: Effects of melancholia, atypical depression, severity of illness, episode number, suicidal ideation and prior suicide attempts. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 372–383. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- van de Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Beaumont, M.; Roura, E.; Lambert, W.; Turni, C.; Michiels, J.; Chalvon-Demersay, T. Selective nourishing of gut microbiota with amino acids: A novel prebiotic approach? Front. Nutr. 2022, 9, 1066898. [Google Scholar] [CrossRef]
- Miri, S.; Yeo, J.; Abubaker, S.; Hammami, R. Neuromicrobiology, an emerging neurometabolic facet of the gut microbiome? Front. Microbiol. 2023, 14, 1098412. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Pereira, J.S.; Rea, K.; Nolan, Y.M.; O’Leary, O.F.; Dinan, T.G.; Cryan, J.F. Depression’s Unholy Trinity: Dysregulated Stress, Immunity, and the Microbiome. Annu. Rev. Psychol. 2020, 71, 49–78. [Google Scholar] [CrossRef] [PubMed]
- Réus, G.Z.; Manosso, L.M.; Quevedo, J.; Carvalho, A.F. Major depressive disorder as a neuro-immune disorder: Origin, mechanisms, and therapeutic opportunities. Neurosci. Biobehav. Rev. 2023, 155, 105425. [Google Scholar] [CrossRef]
- Fülling, C.; Dinan, T.G.; Cryan, J.F. Gut Microbe to Brain Signaling: What Happens in Vagus…. Neuron 2019, 101, 998–1002. [Google Scholar] [CrossRef]
- Siopi, E.; Galerne, M.; Rivagorda, M.; Saha, S.; Moigneu, C.; Moriceau, S.; Bigot, M.; Oury, F.; Lledo, P.M. Gut microbiota changes require vagus nerve integrity to promote depressive-like behaviors in mice. Mol. Psychiatry 2023, 28, 3002–3012. [Google Scholar] [CrossRef]
- Hashimoto, K. Neuroinflammation through the vagus nerve-dependent gut-microbiota-brain axis in treatment-resistant depression. Prog. Brain Res. 2023, 278, 61–77. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, M.; Hao, W.; Wang, Y.; Zhang, T.; Liu, C. Neuroinflammation mechanisms of neuromodulation therapies for anxiety and depression. Transl. Psychiatry 2023, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Donoso, F.; Cryan, J.F.; Olavarría-Ramírez, L.; Nolan, Y.M.; Clarke, G. Inflammation, Lifestyle Factors, and the Microbiome-Gut-Brain Axis: Relevance to Depression and Antidepressant Action. Clin. Pharmacol. Ther. 2023, 113, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Liu, J.; Bonnechere, B.; MahmoudianDehkordi, S.; Arnold, M.; Batra, R.; Chiou, Y.J.; Fernandes, M.; Ikram, M.A.; Kraaij, R.; et al. Interplay of Metabolome and Gut Microbiome in Individuals With Major Depressive Disorder vs Control Individuals. JAMA Psychiatry 2023, 80, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, J.; Liu, P.; Tu, H.; Zhang, R.; Zhang, Y.; Sun, N.; Zhang, K. Gut microbiota composition in depressive disorder: A systematic review, meta-analysis, and meta-regression. Transl. Psychiatry 2023, 13, 379. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jing, K.; Lu, H.; Li, K.; Zhang, Y.; Hasichaolu. Exploring the Correlation between Changes in Gut Microbial Community Diversity and Depression in Human Populations. Biomed. Res. Int. 2022, 2022, 6334868. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gao, M.; Liu, Z.; Zhang, Y.; Tu, H.; Lei, L.; Wu, P.; Zhang, A.; Yang, C.; Li, G.; et al. Gut Microbiome Composition Linked to Inflammatory Factors and Cognitive Functions in First-Episode, Drug-Naive Major Depressive Disorder Patients. Front. Neurosci. 2021, 15, 800764. [Google Scholar] [CrossRef]
- Sun, N.; Zhang, J.; Wang, J.; Liu, Z.; Wang, X.; Kang, P.; Yang, C.; Liu, P.; Zhang, K. Abnormal gut microbiota and bile acids in patients with first-episode major depressive disorder and correlation analysis. Psychiatry Clin. Neurosci. 2022, 76, 321–328. [Google Scholar] [CrossRef]
- Zhang, Q.; Yun, Y.; An, H.; Zhao, W.; Ma, T.; Wang, Z.; Yang, F. Gut microbiome and daytime function in Chinese patients with major depressive disorder. J. Psychosom. Res. 2022, 157, 110787. [Google Scholar] [CrossRef]
- Zhao, H.; Jin, K.; Jiang, C.; Pan, F.; Wu, J.; Luan, H.; Zhao, Z.; Chen, J.; Mou, T.; Wang, Z.; et al. A pilot exploration of multi-omics research of gut microbiome in major depressive disorders. Transl. Psychiatry 2022, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Chen, J.J.; Wang, Y.; Shao, W.H.; Zhou, C.J.; Xie, P. Differential Gut Microbiota Compositions Related With the Severity of Major Depressive Disorder. Front. Cell. Infect. Microbiol. 2022, 12, 907239. [Google Scholar] [CrossRef]
- Maes, M.; Vasupanrajit, A.; Jirakran, K.; Klomkliew, P.; Chanchaem, P.; Tunvirachaisakul, C.; Payungporn, S. Exploration of the Gut Microbiome in Thai Patients with Major Depressive Disorder Shows a Specific Bacterial Profile with Depletion of the. Cells 2023, 12, 1240. [Google Scholar] [CrossRef]
- Liang, S.; Wu, X.; Jin, F. Gut-Brain Psychology: Rethinking Psychology from the Microbiota-Gut-Brain Axis. Front. Integr. Neurosci. 2018, 12, 33. [Google Scholar] [CrossRef]
- Arora, P.; Sagar, R.; Mehta, M.; Pallavi, P.; Sharma, S.; Mukhopadhyay, A.K. Serum S100B levels in patients with depression. Indian. J. Psychiatry 2019, 61, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, Y.; Xia, C.; Lai, Q.; Dong, Z.; Kuang, W.; Yang, C.; Su, D.; Li, H.; Zhong, Z. Potential effects of antibiotic-induced gut microbiome alteration on blood-brain barrier permeability compromise in rhesus monkeys. Ann. N. Y Acad. Sci. 2020, 1470, 14–24. [Google Scholar] [CrossRef]
- Leclercq, S.; Mian, F.M.; Stanisz, A.M.; Bindels, L.B.; Cambier, E.; Ben-Amram, H.; Koren, O.; Forsythe, P.; Bienenstock, J. Low-dose penicillin in early life induces long-term changes in murine gut microbiota, brain cytokines and behavior. Nat. Commun. 2017, 8, 15062. [Google Scholar] [CrossRef]
- Bisgaard, T.H.; Allin, K.H.; Keefer, L.; Ananthakrishnan, A.N.; Jess, T. Depression and anxiety in inflammatory bowel disease: Epidemiology, mechanisms and treatment. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 717–726. [Google Scholar] [CrossRef]
- Maes, M.; Kubera, M.; Leunis, J.C. The gut-brain barrier in major depression: Intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol. Lett. 2008, 29, 117–124. [Google Scholar] [PubMed]
- Takahashi, K.; Nakagawasai, O.; Nemoto, W.; Odaira, T.; Sakuma, W.; Onogi, H.; Nishijima, H.; Furihata, R.; Nemoto, Y.; Iwasa, H.; et al. Effect of Enterococcus faecalis 2001 on colitis and depressive-like behavior in dextran sulfate sodium-treated mice: Involvement of the brain-gut axis. J. Neuroinflamm. 2019, 16, 201. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.W.; Shin, Y.J.; Ma, X.; Son, Y.H.; Jang, H.M.; Lee, C.K.; Kim, D.H. The Alleviation of Gut Microbiota-Induced Depression and Colitis in Mice by Anti-Inflammatory Probiotics NK151, NK173, and NK175. Nutrients 2022, 14, 2080. [Google Scholar] [CrossRef] [PubMed]
- Wadie, W.; Mohamed, S.S.; Abd El-Haleim, E.A.; Khayyal, M.T. Niacin modulates depressive-like behavior in experimental colitis through GPR109A-dependent mechanisms. Life Sci. 2023, 330, 122004. [Google Scholar] [CrossRef]
- Jang, S.E.; Lim, S.M.; Jeong, J.J.; Jang, H.M.; Lee, H.J.; Han, M.J.; Kim, D.H. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018, 11, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; DePierre, J.W.; Nässberger, L. Tricyclic antidepressants inhibit IL-6, IL-1 beta and TNF-alpha release in human blood monocytes and IL-2 and interferon-gamma in T cells. Immunopharmacology 1996, 34, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Zhang, S.; Zong, Y.; Halim, M.; Ren, Z.; Wang, Y.; Ma, Y.; Li, B.; Ma, L.; Zhou, G.; et al. Involvement of the microglial NLRP3 inflammasome in the anti-inflammatory effect of the antidepressant clomipramine. J. Affect. Disord. 2019, 254, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zong, Y.; Ren, Z.; Hu, J.; Wu, X.; Xiao, H.; Qin, S.; Zhou, G.; Ma, Y.; Zhang, Y.; et al. Regulation of indoleamine 2, 3-dioxygenase in hippocampal microglia by NLRP3 inflammasome in lipopolysaccharide-induced depressive-like behaviors. Eur. J. Neurosci. 2020, 52, 4586–4601. [Google Scholar] [CrossRef]
- Faustmann, T.J.; Wawrzyniak, M.; Faustmann, P.M.; Corvace, F.; Ismail, F.S. Inhibition of Microglial Activation by Amitriptyline and Doxepin in Interferon-β Pre-Treated Astrocyte-Microglia Co-Culture Model of Inflammation. Brain Sci. 2023, 13, 493. [Google Scholar] [CrossRef]
- Jeon, S.A.; Lee, E.; Hwang, I.; Han, B.; Park, S.; Son, S.; Yang, J.; Hong, S.; Kim, C.H.; Son, J.; et al. NLRP3 Inflammasome Contributes to Lipopolysaccharide-induced Depressive-like Behaviors via Indoleamine 2,3-dioxygenase Induction. Int. J. Neuropsychopharmacol. 2017, 20, 896–906. [Google Scholar] [CrossRef]
- Maes, M.; Kenis, G.; Kubera, M.; De Baets, M.; Steinbusch, H.; Bosmans, E. The negative immunoregulatory effects of fluoxetine in relation to the cAMP-dependent PKA pathway. Int. Immunopharmacol. 2005, 5, 609–618. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, X.Y.; Zhang, Q.Y.; Kong, L.D. Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain Behav. Immun. 2014, 41, 90–100. [Google Scholar] [CrossRef]
- Tian, M.; Yang, M.; Li, Z.; Wang, Y.; Chen, W.; Yang, L.; Li, Y.; Yuan, H. Fluoxetine suppresses inflammatory reaction in microglia under OGD/R challenge via modulation of NF-κB signaling. Biosci. Rep. 2019, 39, BSR20181584. [Google Scholar] [CrossRef]
- Du, R.H.; Tan, J.; Sun, X.Y.; Lu, M.; Ding, J.H.; Hu, G. Fluoxetine Inhibits NLRP3 Inflammasome Activation: Implication in Depression. Int. J. Neuropsychopharmacol. 2016, 19, pyw037. [Google Scholar] [CrossRef]
- Li, W.; Ali, T.; Zheng, C.; Liu, Z.; He, K.; Shah, F.A.; Ren, Q.; Rahman, S.U.; Li, N.; Yu, Z.J.; et al. Fluoxetine regulates eEF2 activity (phosphorylation) via HDAC1 inhibitory mechanism in an LPS-induced mouse model of depression. J. Neuroinflamm. 2021, 18, 38. [Google Scholar] [CrossRef]
- Fang, Y.; Ding, X.; Zhang, Y.; Cai, L.; Ge, Y.; Ma, K.; Xu, R.; Li, S.; Song, M.; Zhu, H.; et al. Fluoxetine inhibited the activation of A1 reactive astrocyte in a mouse model of major depressive disorder through astrocytic 5-HT2BR/β-arrestin2 pathway. J. Neuroinflamm. 2022, 19, 23. [Google Scholar] [CrossRef]
- Li, J.M.; Liu, L.L.; Su, W.J.; Wang, B.; Zhang, T.; Zhang, Y.; Jiang, C.L. Ketamine may exert antidepressant effects via suppressing NLRP3 inflammasome to upregulate AMPA receptors. Neuropharmacology 2019, 146, 149–153. [Google Scholar] [CrossRef]
- Kopra, E.; Mondelli, V.; Pariante, C.; Nikkheslat, N. Ketamine’s effect on inflammation and kynurenine pathway in depression: A systematic review. J. Psychopharmacol. 2021, 35, 934–945. [Google Scholar] [CrossRef]
- Chen, M.H.; Li, C.T.; Lin, W.C.; Hong, C.J.; Tu, P.C.; Bai, Y.M.; Cheng, C.M.; Su, T.P. Rapid inflammation modulation and antidepressant efficacy of a low-dose ketamine infusion in treatment-resistant depression: A randomized, double-blind control study. Psychiatry Res. 2018, 269, 207–211. [Google Scholar] [CrossRef]
- Kiraly, D.D.; Horn, S.R.; Van Dam, N.T.; Costi, S.; Schwartz, J.; Kim-Schulze, S.; Patel, M.; Hodes, G.E.; Russo, S.J.; Merad, M.; et al. Altered peripheral immune profiles in treatment-resistant depression: Response to ketamine and prediction of treatment outcome. Transl. Psychiatry 2017, 7, e1065. [Google Scholar] [CrossRef]
- Kopschina Feltes, P.; Doorduin, J.; Klein, H.C.; Juárez-Orozco, L.E.; Dierckx, R.A.; Moriguchi-Jeckel, C.M.; de Vries, E.F. Anti-inflammatory treatment for major depressive disorder: Implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J. Psychopharmacol. 2017, 31, 1149–1165. [Google Scholar] [CrossRef]
- Guo, J.Y.; Li, C.Y.; Ruan, Y.P.; Sun, M.; Qi, X.L.; Zhao, B.S.; Luo, F. Chronic treatment with celecoxib reverses chronic unpredictable stress-induced depressive-like behavior via reducing cyclooxygenase-2 expression in rat brain. Eur. J. Pharmacol. 2009, 612, 54–60. [Google Scholar] [CrossRef]
- Kurhe, Y.; Mahesh, R.; Gupta, D. Effect of a selective cyclooxygenase type 2 inhibitor celecoxib on depression associated with obesity in mice: An approach using behavioral tests. Neurochem. Res. 2014, 39, 1395–1402. [Google Scholar] [CrossRef]
- Maciel, I.S.; Silva, R.B.; Morrone, F.B.; Calixto, J.B.; Campos, M.M. Synergistic effects of celecoxib and bupropion in a model of chronic inflammation-related depression in mice. PLoS ONE 2013, 8, e77227. [Google Scholar] [CrossRef]
- Dinur, E.; Goldenberg, H.; Robinson, E.; Naggan, L.; Kozela, E.; Yirmiya, R. A Novel Anti-Inflammatory Formulation Comprising Celecoxib and Cannabidiol Exerts Antidepressant and Anxiolytic Effects. Cannabis Cannabinoid Res. 2022. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Q.; Wang, Q. Effect of celecoxib on improving depression: A systematic review and meta-analysis. World J. Clin. Cases 2022, 10, 7872–7882. [Google Scholar] [CrossRef]
- Gędek, A.; Szular, Z.; Antosik, A.Z.; Mierzejewski, P.; Dominiak, M. Celecoxib for Mood Disorders: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Clin. Med. 2023, 12, 3497. [Google Scholar] [CrossRef]
- Seo, M.K.; Lee, J.G.; Park, S.W. Effects of escitalopram and ibuprofen on a depression-like phenotype induced by chronic stress in rats. Neurosci. Lett. 2019, 696, 168–173. [Google Scholar] [CrossRef]
- Salmani, H.; Hosseini, M.; Baghcheghi, Y.; Samadi-Noshahr, Z. The brain consequences of systemic inflammation were not fully alleviated by ibuprofen treatment in mice. Pharmacol. Rep. 2021, 73, 130–142. [Google Scholar] [CrossRef]
- Warner-Schmidt, J.L.; Vanover, K.E.; Chen, E.Y.; Marshall, J.J.; Greengard, P. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 9262–9267. [Google Scholar] [CrossRef]
- Brunello, N.; Alboni, S.; Capone, G.; Benatti, C.; Blom, J.M.; Tascedda, F.; Kriwin, P.; Mendlewicz, J. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int. Clin. Psychopharmacol. 2006, 21, 219–225. [Google Scholar] [CrossRef]
- Alboni, S.; Benatti, C.; Capone, G.; Tascedda, F.; Brunello, N. Neither all anti-inflammatory drugs nor all doses are effective in accelerating the antidepressant-like effect of fluoxetine in an animal model of depression. J. Affect. Disord. 2018, 235, 124–128. [Google Scholar] [CrossRef]
- Dominiak, M.; Gędek, A.; Sikorska, M.; Mierzejewski, P.; Wojnar, M.; Antosik-Wójcińska, A.Z. Acetylsalicylic Acid and Mood Disorders: A Systematic Review. Pharmaceuticals 2022, 16, 67. [Google Scholar] [CrossRef]
- Souza, P.B.; de Araujo Borba, L.; Castro de Jesus, L.; Valverde, A.P.; Gil-Mohapel, J.; Rodrigues, A.L.S. Major Depressive Disorder and Gut Microbiota: Role of Physical Exercise. Int. J. Mol. Sci. 2023, 24, 16870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ding, S.; Wang, R. Research Progress of Mitochondrial Mechanism in NLRP3 Inflammasome Activation and Exercise Regulation of NLRP3 Inflammasome. Int. J. Mol. Sci. 2021, 22, 866. [Google Scholar] [CrossRef]
- Tang, X.Q.; Liao, R.Y.; Zheng, L.J.; Yang, L.L.; Ma, Z.L.; Yi, C.; Liu, J.; Liu, J.C.; Kuang, Y.J.; Cai, H.A.; et al. Aerobic exercise reverses the NF-κB/NLRP3 inflammasome/5-HT pathway by upregulating irisin to alleviate post-stroke depression. Ann. Transl. Med. 2022, 10, 1350. [Google Scholar] [CrossRef]
- Li, C.; Xu, X.; Wang, Z.; Wang, Y.; Luo, L.; Cheng, J.; Chen, S.F.; Liu, H.; Wan, Q.; Wang, Q. Exercise ameliorates post-stroke depression by inhibiting PTEN elevation-mediated upregulation of TLR4/NF-κB/NLRP3 signaling in mice. Brain Res. 2020, 1736, 146777. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, H.; Qu, H.; Chen, Y.; Bai, Q.; Guo, F.; Wang, L.; Jiang, X.; Mao, H. Effects of aerobic exercise on depression-like behavior and TLR4/NLRP3 pathway in hippocampus CA1 region of CUMS-depressed mice. J. Affect. Disord. 2023, 341, 248–255. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Sheng, H.; Ni, X.; Lu, J. Exercise amelioration of depression-like behavior in OVX mice is associated with suppression of NLRP3 inflammasome activation in hippocampus. Behav. Brain Res. 2016, 307, 18–24. [Google Scholar] [CrossRef]
- Rosa, J.M.; Camargo, A.; Wolin, I.A.V.; Kaster, M.P.; Rodrigues, A.L.S. Physical exercise prevents amyloid β1-40-induced disturbances in NLRP3 inflammasome pathway in the hippocampus of mice. Metab. Brain Dis. 2021, 36, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Xu, T. Aerobic Exercise Improves Depressive-like Behavior in CUMS-Induced Rats via the SIRT3/ROS/NLRP3 Signaling Pathway. Life 2023, 13, 1711. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wang, H.; Li, J.J.; Zhang, Y.L.; Xin, L.; Li, F.; Lou, S.J. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 2016, 57, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, J.; Liu, Y.; Li, J.; Liu, B.; Li, M.; Lou, S. Aerobic Exercise Improves Synaptic-Related Proteins of Diabetic Rats by Inhibiting FOXO1/NF-κB/NLRP3 Inflammatory Signaling Pathway and Ameliorating PI3K/Akt Insulin Signaling Pathway. J. Mol. Neurosci. 2019, 69, 28–38. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Liu, B.; Li, F.; Hu, J.; Wang, Q.; Li, M.; Lou, S. Mechanisms of Aerobic Exercise Upregulating the Expression of Hippocampal Synaptic Plasticity-Associated Proteins in Diabetic Rats. Neural Plast. 2019, 2019, 7920540. [Google Scholar] [CrossRef]
- Ding, Y.; Xu, X. Effects of regular exercise on inflammasome activation-related inflammatory cytokine levels in older adults: A systematic review and meta-analysis. J. Sports Sci. 2021, 39, 2338–2352. [Google Scholar] [CrossRef]
- Comassi, M.; Santini, E.; Rossi, C.; Vitolo, E.; Seghieri, M.; Tocchini, L.; Franzoni, F.; Solini, A. The level of physical training modulates cytokine levels through P2X7 receptor in healthy subjects. Eur. J. Clin. Investig. 2018, 48, e12880. [Google Scholar] [CrossRef]
- Khakroo Abkenar, I.; Rahmani-Nia, F.; Lombardi, G. The Effects of Acute and Chronic Aerobic Activity on the Signaling Pathway of the Inflammasome NLRP3 Complex in Young Men. Medicina 2019, 55, 105. [Google Scholar] [CrossRef] [PubMed]
- Rohnejad, B.; Monazzami, A. Effects of high-intensity intermittent training on some inflammatory and muscle damage indices in overweight middle-aged men. Apunt. Sports Med. 2023, 58, 9. [Google Scholar] [CrossRef]
- Cerqueira, É.; Marinho, D.A.; Neiva, H.P.; Lourenço, O. Inflammatory Effects of High and Moderate Intensity Exercise—A Systematic Review. Front. Physiol. 2019, 10, 1550. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Gao, L.; Li, T.; Shao, A.; Zhang, J. Neuroprotective Role of Agmatine in Neurological Diseases. Curr. Neuropharmacol. 2018, 16, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Olescowicz, G.; Neis, V.B.; Fraga, D.B.; Rosa, P.B.; Azevedo, D.P.; Melleu, F.F.; Brocardo, P.S.; Gil-Mohapel, J.; Rodrigues, A.L.S. Antidepressant and pro-neurogenic effects of agmatine in a mouse model of stress induced by chronic exposure to corticosterone. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.E.; Egea, J.; Buendia, I.; Gómez-Rangel, V.; Parada, E.; Navarro, E.; Casas, A.I.; Wojnicz, A.; Ortiz, J.A.; Cuadrado, A.; et al. Agmatine, by Improving Neuroplasticity Markers and Inducing Nrf2, Prevents Corticosterone-Induced Depressive-like Behavior in Mice. Mol. Neurobiol. 2016, 53, 3030–3045. [Google Scholar] [CrossRef] [PubMed]
- Gawali, N.B.; Bulani, V.D.; Gursahani, M.S.; Deshpande, P.S.; Kothavade, P.S.; Juvekar, A.R. Agmatine attenuates chronic unpredictable mild stress-induced anxiety, depression-like behaviours and cognitive impairment by modulating nitrergic signalling pathway. Brain Res. 2017, 1663, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Neis, V.B.; Bettio, L.E.B.; Moretti, M.; Rosa, P.B.; Ribeiro, C.M.; Freitas, A.E.; Gonçalves, F.M.; Leal, R.B.; Rodrigues, A.L.S. Acute agmatine administration, similar to ketamine, reverses depressive-like behavior induced by chronic unpredictable stress in mice. Pharmacol. Biochem. Behav. 2016, 150–151, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.E.; Bettio, L.E.; Neis, V.B.; Santos, D.B.; Ribeiro, C.M.; Rosa, P.B.; Farina, M.; Rodrigues, A.L. Agmatine abolishes restraint stress-induced depressive-like behavior and hippocampal antioxidant imbalance in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 50, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.D.; Chen, W.Q.; Wang, Z.Y.; Cao, D.N.; Wu, N.; Li, J. Antidepressant-like action of agmatine in the acute and sub-acute mouse models of depression: A receptor mechanism study. Metab. Brain Dis. 2018, 33, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Sahin Ozkartal, C.; Tuzun, E.; Kucukali, C.I.; Ulusoy, C.; Giris, M.; Aricioglu, F. Antidepressant-like effects of agmatine and NOS inhibitors in chronic unpredictable mild stress model of depression in rats: The involvement of NLRP inflammasomes. Brain Res. 2019, 1725, 146438. [Google Scholar] [CrossRef]
- Gawali, N.B.; Bulani, V.D.; Chowdhury, A.A.; Deshpande, P.S.; Nagmoti, D.M.; Juvekar, A.R. Agmatine ameliorates lipopolysaccharide induced depressive-like behaviour in mice by targeting the underlying inflammatory and oxido-nitrosative mediators. Pharmacol. Biochem. Behav. 2016, 149, 1–8. [Google Scholar] [CrossRef]
- Neis, V.B.; Manosso, L.M.; Moretti, M.; Freitas, A.E.; Daufenbach, J.; Rodrigues, A.L. Depressive-like behavior induced by tumor necrosis factor-α is abolished by agmatine administration. Behav. Brain Res. 2014, 261, 336–344. [Google Scholar] [CrossRef]
- Sahin, C.; Albayrak, O.; Akdeniz, T.F.; Akbulut, Z.; Yanikkaya Demirel, G.; Aricioglu, F. Agmatine Reverses Sub-chronic Stress induced Nod-like Receptor Protein 3 (NLRP3) Activation and Cytokine Response in Rats. Basic Clin. Pharmacol. Toxicol. 2016, 119, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, J.; Hua, Y.; Gong, J.; Ding, S.; Du, Y.; Wang, X.; Zheng, R.; Xu, H. Agmatine Alleviates Epileptic Seizures and Hippocampal Neuronal Damage by Inhibiting Gasdermin D-Mediated Pyroptosis. Front. Pharmacol. 2021, 12, 627557. [Google Scholar] [CrossRef]
- Kale, M.; Nimje, N.; Aglawe, M.M.; Umekar, M.; Taksande, B.; Kotagale, N. Agmatine modulates anxiety and depression-like behaviour in diabetic insulin-resistant rats. Brain Res. 2020, 1747, 147045. [Google Scholar] [CrossRef]
- Kotagale, N.; Rahangdale, S.; Borkar, A.; Singh, K.; Ikhar, A.; Takale, N.; Umekar, M.; Taksande, B. Possible involvement of agmatine in neuropharmacological actions of metformin in diabetic mice. Eur. J. Pharmacol. 2021, 907, 174255. [Google Scholar] [CrossRef] [PubMed]
- Azar, Y.O.; Badawi, G.A.; Zaki, H.F.; Ibrahim, S.M. Agmatine-mediated inhibition of NMDA receptor expression and amelioration of dyskinesia via activation of Nrf2 and suppression of HMGB1/RAGE/TLR4/MYD88/NF-κB signaling cascade in rotenone lesioned rats. Life Sci. 2022, 311 Pt A, 121049. [Google Scholar] [CrossRef]
- El-Sayed, E.K.; Ahmed, A.; Morsy, E.E.; Nofal, S. Neuroprotective effect of agmatine (decarboxylated l-arginine) against oxidative stress and neuroinflammation in rotenone model of Parkinson’s disease. Hum. Exp. Toxicol. 2019, 38, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sim, A.Y.; Barua, S.; Kim, J.Y.; Lee, J.E. Agmatine-IRF2BP2 interaction induces M2 phenotype of microglia by increasing IRF2-KLF4 signaling. Inflamm. Res. 2023, 72, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, K.; Stevanovic, I.; Bozic, I.D.; Milosevic, A.; Janjic, M.M.; Laketa, D.; Bjelobaba, I.; Lavrnja, I.; Savic, D. Agmatine Mitigates Inflammation-Related Oxidative Stress in BV-2 Cells by Inducing a Pre-Adaptive Response. Int. J. Mol. Sci. 2022, 23, 105. [Google Scholar] [CrossRef]
- Song, J.; Lee, B.; Kang, S.; Oh, Y.; Kim, E.; Kim, C.H.; Song, H.T.; Lee, J.E. Agmatine Ameliorates High Glucose-Induced Neuronal Cell Senescence by Regulating the p21 and p53 Signaling. Exp. Neurobiol. 2016, 25, 24–32. [Google Scholar] [CrossRef]
- Sabit, H.; Kassab, A.; Alaa, D.; Mohamed, S.; Abdel-Ghany, S.; Mansy, M.; Said, O.A.; Khalifa, M.A.; Hafiz, H.; Abushady, A.M. The Effect of Probiotic Supplementation on the Gut-Brain Axis in Psychiatric Patients. Curr. Issues Mol. Biol. 2023, 45, 4080–4099. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Halloran, K.; Underwood, M.A. Probiotic mechanisms of action. Early Hum. Dev. 2019, 135, 58–65. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, J.P.; Deng, K.; Li, X.; Yuan, Y.; Xuan, Q.; Xie, J.; He, X.M.; Wang, Q.; Li, J.J.; et al. Prophylactic Effects of Bifidobacterium adolescentis on Anxiety and Depression-like Phenotypes after Chronic Stress: A Role of the Gut Microbiota-Inflammation Axis. Front. Behav. Neurosci. 2019, 13, 126. [Google Scholar] [CrossRef]
- Tian, T.; Xu, B.; Qin, Y.; Fan, L.; Chen, J.; Zheng, P.; Gong, X.; Wang, H.; Bai, M.; Pu, J.; et al. Clostridium butyricum miyairi 588 has preventive effects on chronic social defeat stress-induced depressive-like behaviour and modulates microglial activation in mice. Biochem. Biophys. Res. Commun. 2019, 516, 430–436. [Google Scholar] [CrossRef]
- Akkasheh, G.; Kashani-Poor, Z.; Tajabadi-Ebrahimi, M.; Jafari, P.; Akbari, H.; Taghizadeh, M.; Memarzadeh, M.R.; Asemi, Z.; Esmaillzadeh, A. Clinical and metabolic response to probiotic administration in patients with major depressive disorder: A randomized, double-blind, placebo-controlled trial. Nutrition 2016, 32, 315–320. [Google Scholar] [CrossRef]
- Kazemi, A.; Noorbala, A.A.; Azam, K.; Eskandari, M.H.; Djafarian, K. Effect of probiotic and prebiotic vs placebo on psychological outcomes in patients with major depressive disorder: A randomized clinical trial. Clin. Nutr. 2019, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C-Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Camargo, A.; Rodrigues, A.L.S. Novel Targets for Fast Antidepressant Responses: Possible Role of Endogenous Neuromodulators. Chronic Stress 2019, 3, 2470547019858083. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Ascorbic Acid to Manage Psychiatric Disorders. CNS Drugs 2017, 31, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J. On the effect of vitamin C intake on human health: How to (mis)interprete the clinical evidence. Redox Biol. 2020, 34, 101532. [Google Scholar] [CrossRef]
- Nualart, F.; Mack, L.; García, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martínez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef]
- Chang, C.W.; Chen, M.J.; Wang, T.E.; Chang, W.H.; Lin, C.C.; Liu, C.Y. Scurvy in a patient with depression. Dig. Dis. Sci. 2007, 52, 1259–1261. [Google Scholar] [CrossRef]
- Plevin, D.; Galletly, C. The neuropsychiatric effects of vitamin C deficiency: A systematic review. BMC Psychiatry 2020, 20, 315. [Google Scholar] [CrossRef]
- Gariballa, S. Poor vitamin C status is associated with increased depression symptoms following acute illness in older people. Int. J. Vitam. Nutr. Res. 2014, 84, 12–17. [Google Scholar] [CrossRef]
- Binfaré, R.W.; Rosa, A.O.; Lobato, K.R.; Santos, A.R.; Rodrigues, A.L. Ascorbic acid administration produces an antidepressant-like effect: Evidence for the involvement of monoaminergic neurotransmission. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 530–540. [Google Scholar] [CrossRef]
- Moretti, M.; Colla, A.; de Oliveira Balen, G.; dos Santos, D.B.; Budni, J.; de Freitas, A.E.; Farina, M.; Severo Rodrigues, A.L. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J. Psychiatr. Res. 2012, 46, 331–340. [Google Scholar] [CrossRef]
- Moretti, M.; Budni, J.; Freitas, A.E.; Rosa, P.B.; Rodrigues, A.L. Antidepressant-like effect of ascorbic acid is associated with the modulation of mammalian target of rapamycin pathway. J. Psychiatr. Res. 2014, 48, 16–24. [Google Scholar] [CrossRef]
- Moretti, M.; Budni, J.; Ribeiro, C.M.; Rieger, D.K.; Leal, R.B.; Rodrigues, A.L.S. Subchronic administration of ascorbic acid elicits antidepressant-like effect and modulates cell survival signaling pathways in mice. J. Nutr. Biochem. 2016, 38, 50–56. [Google Scholar] [CrossRef]
- Maratha, S.; Sharma, V.; Walia, V. Antidepressant Like Effect of Ascorbic Acid in Mice: Possible Involvement of NO-sGC-cGMP Signaling. Neurochem. Res. 2022, 47, 967–978. [Google Scholar] [CrossRef]
- Moretti, M.; Budni, J.; Freitas, A.E.; Neis, V.B.; Ribeiro, C.M.; de Oliveira Balen, G.; Rieger, D.K.; Leal, R.B.; Rodrigues, A.L. TNF-α-induced depressive-like phenotype and p38(MAPK) activation are abolished by ascorbic acid treatment. Eur. Neuropsychopharmacol. 2015, 25, 902–912. [Google Scholar] [CrossRef]
- Shivavedi, N.; Kumar, M.; Tej, G.N.V.C.; Nayak, P.K. Metformin and ascorbic acid combination therapy ameliorates type 2 diabetes mellitus and comorbid depression in rats. Brain Res. 2017, 1674, 1–9. [Google Scholar] [CrossRef]
- da Silva Souza, S.V.; da Rosa, P.B.; Neis, V.B.; Moreira, J.D.; Rodrigues, A.L.S.; Moretti, M. Effects of cholecalciferol on behavior and production of reactive oxygen species in female mice subjected to corticosterone-induced model of depression. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 111–120. [Google Scholar] [CrossRef]
- Fedotova, J.; Dudnichenko, T.; Kruzliak, P.; Puchavskaya, Z. Different effects of vitamin D hormone treatment on depression-like behavior in the adult ovariectomized female rats. Biomed. Pharmacother. 2016, 84, 1865–1872. [Google Scholar] [CrossRef]
- Neis, V.B.; Werle, I.; Moretti, M.; Rosa, P.B.; Camargo, A.; de O. Dalsenter, Y.; Platt, N.; Rosado, A.F.; Engel, W.D.; de Almeida, G.R.L.; et al. Involvement of serotonergic neurotransmission in the antidepressant-like effect elicited by cholecalciferol in the chronic unpredictable stress model in mice. Metab. Brain Dis. 2022, 37, 1597–1608. [Google Scholar] [CrossRef]
- Kouba, B.R.; Torrá, A.C.N.C.; Camargo, A.; Rodrigues, A.L.S. The antidepressant-like effect elicited by vitamin D. Metab. Brain Dis. 2023, 38, 601–611. [Google Scholar] [CrossRef]
- Camargo, A.; Dalmagro, A.P.; Platt, N.; F Rosado, A.; B Neis, V.; B Zeni, A.L.; P Kaster, M.; S Rodrigues, A.L. Cholecalciferol abolishes depressive-like behavior and hippocampal glucocorticoid receptor impairment induced by chronic corticosterone administration in mice. Pharmacol. Biochem. Behav. 2020, 196, 172971. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Guo, Y.J.; Wang, K.Y.; Chen, L.M.; Jiang, P. Neuroprotective effects of vitamin D and 17ß-estradiol against ovariectomy-induced neuroinflammation and depressive-like state: Role of the AMPK/NF-κB pathway. Int. Immunopharmacol. 2020, 86, 106734. [Google Scholar] [CrossRef]
- Sassi, F.; Tamone, C.; D’Amelio, P. Vitamin D: Nutrient, Hormone, and Immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef]
- Kouba, B.R.; Camargo, A.; Rodrigues, A.L.S. Neuroinflammation in Alzheimer’s disease: Potential beneficial effects of vitamin D. Metab. Brain Dis. 2023, 38, 819–829. [Google Scholar] [CrossRef]
- Baeke, F.; Takiishi, T.; Korf, H.; Gysemans, C.; Mathieu, C. Vitamin D: Modulator of the immune system. Curr. Opin. Pharmacol. 2010, 10, 482–496. [Google Scholar] [CrossRef]
- Cohen-Lahav, M.; Shany, S.; Tobvin, D.; Chaimovitz, C.; Douvdevani, A. Vitamin D decreases NFkappaB activity by increasing IkappaBalpha levels. Nephrol. Dial. Transplant. 2006, 21, 889–897. [Google Scholar] [CrossRef]
- Verma, R.; Jung, J.H.; Kim, J.Y. 1,25-Dihydroxyvitamin D3 up-regulates TLR10 while down-regulating TLR2, 4, and 5 in human monocyte THP-1. J. Steroid Biochem. Mol. Biol. 2014, 141, 1–6. [Google Scholar] [CrossRef]
- Cui, C.; Xu, P.; Li, G.; Qiao, Y.; Han, W.; Geng, C.; Liao, D.; Yang, M.; Chen, D.; Jiang, P. Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: Role of renin-angiotensin system. Redox Biol. 2019, 26, 101295. [Google Scholar] [CrossRef]
- Xin, L.; Che, B.; Zhai, B.; Luo, Q.; Zhang, C.; Wang, J.; Wang, S.; Fan, G.; Liu, Z.; Feng, J.; et al. 1,25-Dihydroxy Vitamin D3 Attenuates the Oxidative Stress-Mediated Inflammation Induced by PM2.5via the p38/NF-κB/NLRP3 Pathway. Inflammation 2019, 42, 702–713. [Google Scholar] [CrossRef]
- Flanagan, P.K.; Chiewchengchol, D.; Wright, H.L.; Edwards, S.W.; Alswied, A.; Satsangi, J.; Subramanian, S.; Rhodes, J.M.; Campbell, B.J. Killing of Escherichia coli by Crohn’s Disease Monocyte-derived Macrophages and Its Enhancement by Hydroxychloroquine and Vitamin D. Inflamm. Bowel Dis. 2015, 21, 1499–1510. [Google Scholar] [CrossRef]
- Du, J.; Chen, Y.; Shi, Y.; Liu, T.; Cao, Y.; Tang, Y.; Ge, X.; Nie, H.; Zheng, C.; Li, Y.C. 1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway. Inflamm. Bowel Dis. 2015, 21, 2495–2506. [Google Scholar] [CrossRef]
- Fakhoury, H.M.A.; Kvietys, P.R.; AlKattan, W.; Anouti, F.A.; Elahi, M.A.; Karras, S.N.; Grant, W.B. Vitamin D and intestinal homeostasis: Barrier, microbiota, and immune modulation. J. Steroid Biochem. Mol. Biol. 2020, 200, 105663. [Google Scholar] [CrossRef]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav. Immun. 2020, 87, 901–909. [Google Scholar] [CrossRef]
- Gold, S.M.; Köhler-Forsberg, O.; Moss-Morris, R.; Mehnert, A.; Miranda, J.J.; Bullinger, M.; Steptoe, A.; Whooley, M.A.; Otte, C. Comorbid depression in medical diseases. Nat. Rev. Dis. Primers 2020, 6, 69. [Google Scholar] [CrossRef]



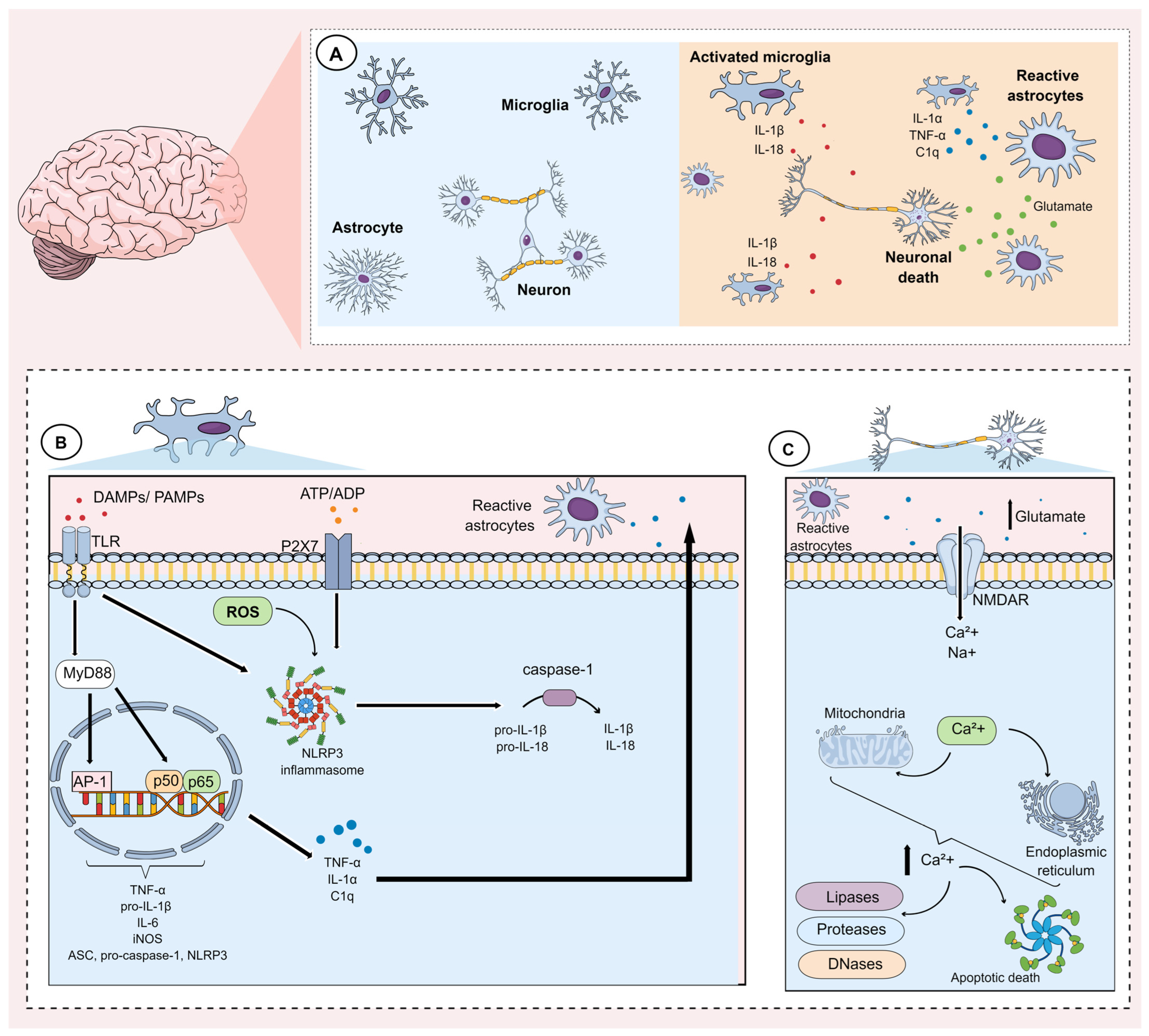{kind=link}
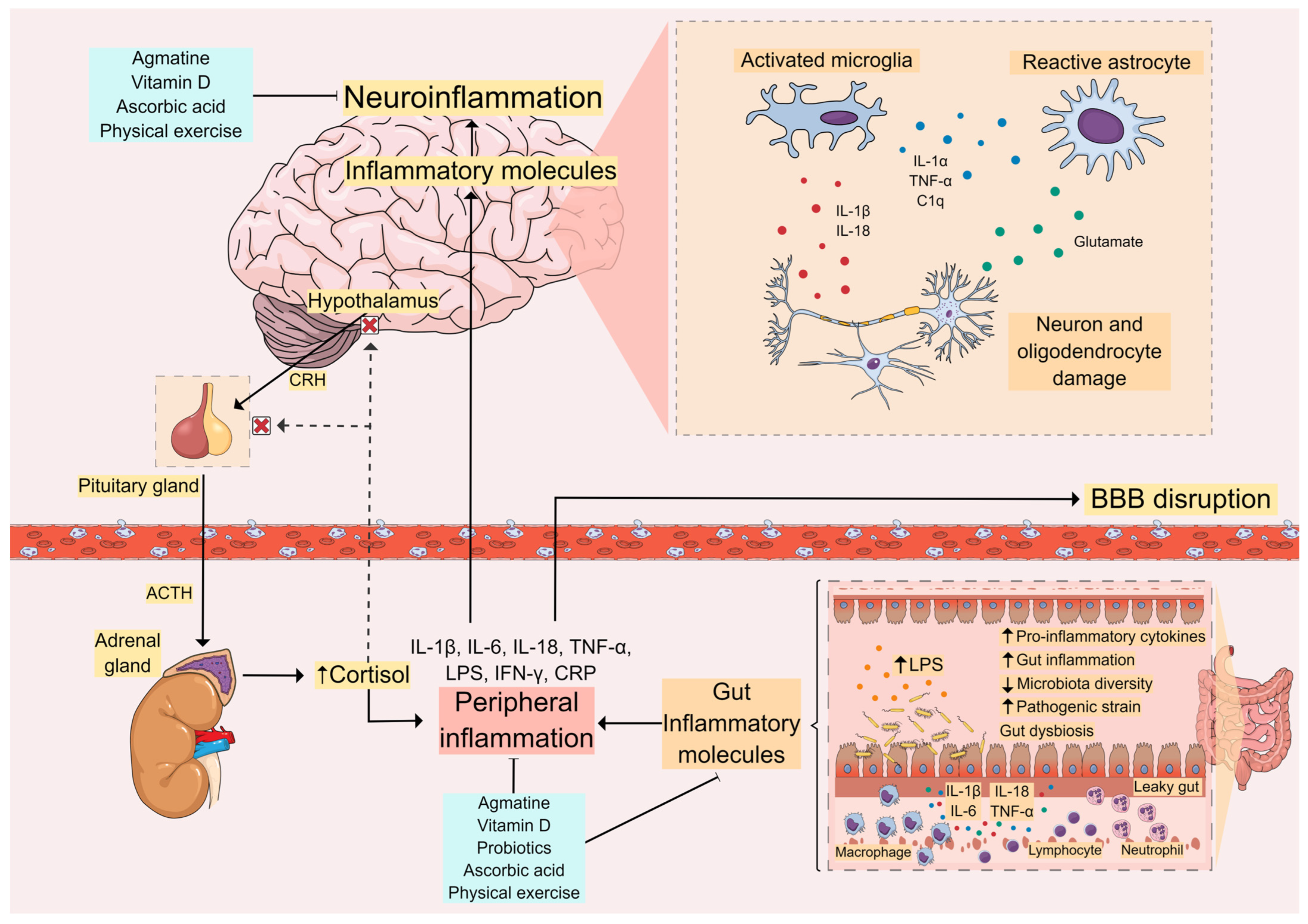{kind=link}
| Biomarker | Animal Model | Sample | Studies |
|---|---|---|---|
| ↑ IL-1β | Mice submitted to chronic stress | Hippocampus | [33] |
| Hippocampus | [45] | ||
| Serum | [238] | ||
| Hippocampus | [262] | ||
| Hippocampus | [265] | ||
| Hippocampus | [296] | ||
| Rats submitted to chronic stress | Cortex | [32] | |
| Prefrontal cortex | [236] | ||
| Prefrontal cortex | [281] | ||
| Mice submitted to chronic social defeat stress | Hippocampus and colon | [297] | |
| LPS in mice | Hippocampus | [45] | |
| Hippocampus | [231] | ||
| Serum and hippocampus | [239] | ||
| Hippocampus | [241] | ||
| Fecal microbiota transplantation from inflammatory bowel disease patients, to mice | Blood and colon | [227] | |
| Ovariectomized mice | Hippocampus | [263] | |
| Rats submitted to an acute restraint stress | Serum, hippocampus, and prefrontal cortex | [284] | |
| Mice submitted to streptozotocin-induced type-II diabetes mellitus | Prefrontal cortex and hippocampus | [287] | |
| ↑ IL-18 | Mice submitted to chronic stress | Hippocampus | [265] |
| Ovariectomized mice | Hippocampus | [263] | |
| Rats submitted to an acute restraint stress | Serum, hippocampus, and prefrontal cortex | [284] | |
| ↑ IL-6 | Mice submitted to chronic stress | Hippocampus | [34] |
| Hippocampus | [33] | ||
| Hippocampus | [45] | ||
| Rats submitted to chronic stress | Hypothalamus and spleen | [32] | |
| Mice submitted to chronic social defeat stress | Hippocampus and colon | [237] | |
| LPS in mice | Hippocampus | [45] | |
| Hippocampus | [231] | ||
| Serum and hippocampus | [239] | ||
| Fecal microbiota transplantation from inflammatory bowel disease patients, to mice | Blood and colon | [227] | |
| Mice with inflammatory bowel disease induced by dextran sulfate sodium (DSS) | Rectum and hippocampus | [226] | |
| Rats submitted to high-fat diet and streptozotocin-induced type-II diabetes mellitus | Hippocampus | [286] | |
| Mice submitted to streptozotocin-induced type-II diabetes mellitus | Prefrontal cortex and hippocampus | [287] | |
| Rat model of type 2 diabetes mellitus and comorbid depression | Prefrontal cortex | [314] | |
| ↑ TNF-α | Mice submitted to chronic stress | Hippocampus | [33] |
| Hippocampus | [45] | ||
| Hippocampus | [278] | ||
| Rats submitted to chronic stress | Hippocampus, cortex and spleen | [32] | |
| LPS in mice | Hippocampus | [45] | |
| Hippocampus | [222] | ||
| Serum and hippocampus | [230] | ||
| Mice with inflammatory bowel disease induced by dextran sulfate sodium (DSS) | Rectum and hippocampus | [217] | |
| Rats submitted to high-fat diet and streptozotocin-induced type-II diabetes mellitus | Hippocampus | [277] | |
| Mice submitted to streptozotocin-induced type-II diabetes mellitus | Prefrontal cortex and hippocampus | [287] | |
| Rat model of type 2 diabetes mellitus and comorbid depression | Prefrontal cortex | [314] |
| Biomarker | Study Design | Subjects | Sample | Studies |
|---|---|---|---|---|
| ↑ IL-1β | Cross-sectional | Adults, 130 MDD and 40 HC | Blood | [28] |
| Longitudinal | Young adults, 50 MDD treatment-naïve and 50 HC | Plasma | [183] | |
| ↑ IL-6 | Longitudinal | Young adults, 50 MDD treatment-naïve and 50 HC | Plasma | [183] |
| Longitudinal | Adolescents, 201 volunteered | Plasma | [182] | |
| Case-Control | Adolescent, 77 MDD and 54 HC | Serum | [180] | |
| ↑ CRP | Longitudinal | Young adults, 50 MDD treatment-naïve and 50 HC | Plasma | [183] |
| Cross-sectional | Adults, 811 MDD | Serum | [181] | |
| Longitudinal | Adolescents, 201 volunteered | Plasma | [182] | |
| ↑ IFN-γ | Cross-sectional | Adults, 103 MDD and 97 HC | Blood | [179] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouba, B.R.; de Araujo Borba, L.; Borges de Souza, P.; Gil-Mohapel, J.; Rodrigues, A.L.S. Role of Inflammatory Mechanisms in Major Depressive Disorder: From Etiology to Potential Pharmacological Targets. Cells 2024, 13, 423. https://doi.org/10.3390/cells13050423
Kouba BR, de Araujo Borba L, Borges de Souza P, Gil-Mohapel J, Rodrigues ALS. Role of Inflammatory Mechanisms in Major Depressive Disorder: From Etiology to Potential Pharmacological Targets. Cells. 2024; 13(5):423. https://doi.org/10.3390/cells13050423
Chicago/Turabian StyleKouba, Bruna R., Laura de Araujo Borba, Pedro Borges de Souza, Joana Gil-Mohapel, and Ana Lúcia S. Rodrigues. 2024. "Role of Inflammatory Mechanisms in Major Depressive Disorder: From Etiology to Potential Pharmacological Targets" Cells 13, no. 5: 423. https://doi.org/10.3390/cells13050423
APA StyleKouba, B. R., de Araujo Borba, L., Borges de Souza, P., Gil-Mohapel, J., & Rodrigues, A. L. S. (2024). Role of Inflammatory Mechanisms in Major Depressive Disorder: From Etiology to Potential Pharmacological Targets. Cells, 13(5), 423. https://doi.org/10.3390/cells13050423