.png)
Cancer Drug Resistance: Targeting Proliferation or Programmed Cell Death

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
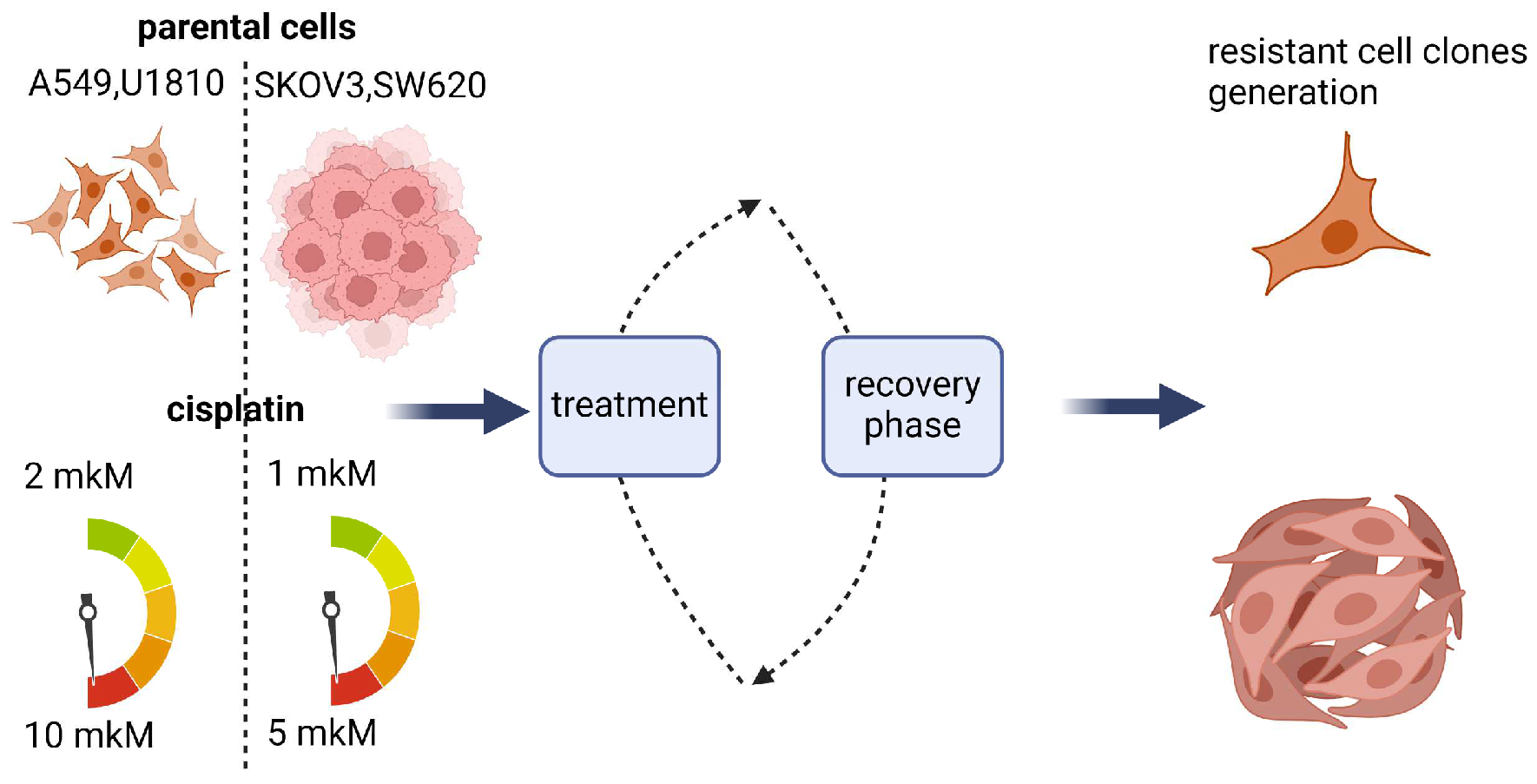
2.2. Development of Drug-Resistant Cell Lines
2.3. Drug Sensitivity Assay
2.4. LIVE/DEAD Assay for Mammalian Cells
2.5. Western Blotting
2.6. Clonogenic Assay
2.7. Metabolic Assays
2.8. Flow Cytometry Analysis of subG1 Population and Cell Death Evaluation
2.9. Data Processing and Statistical Analysis
3. Results and Discussion
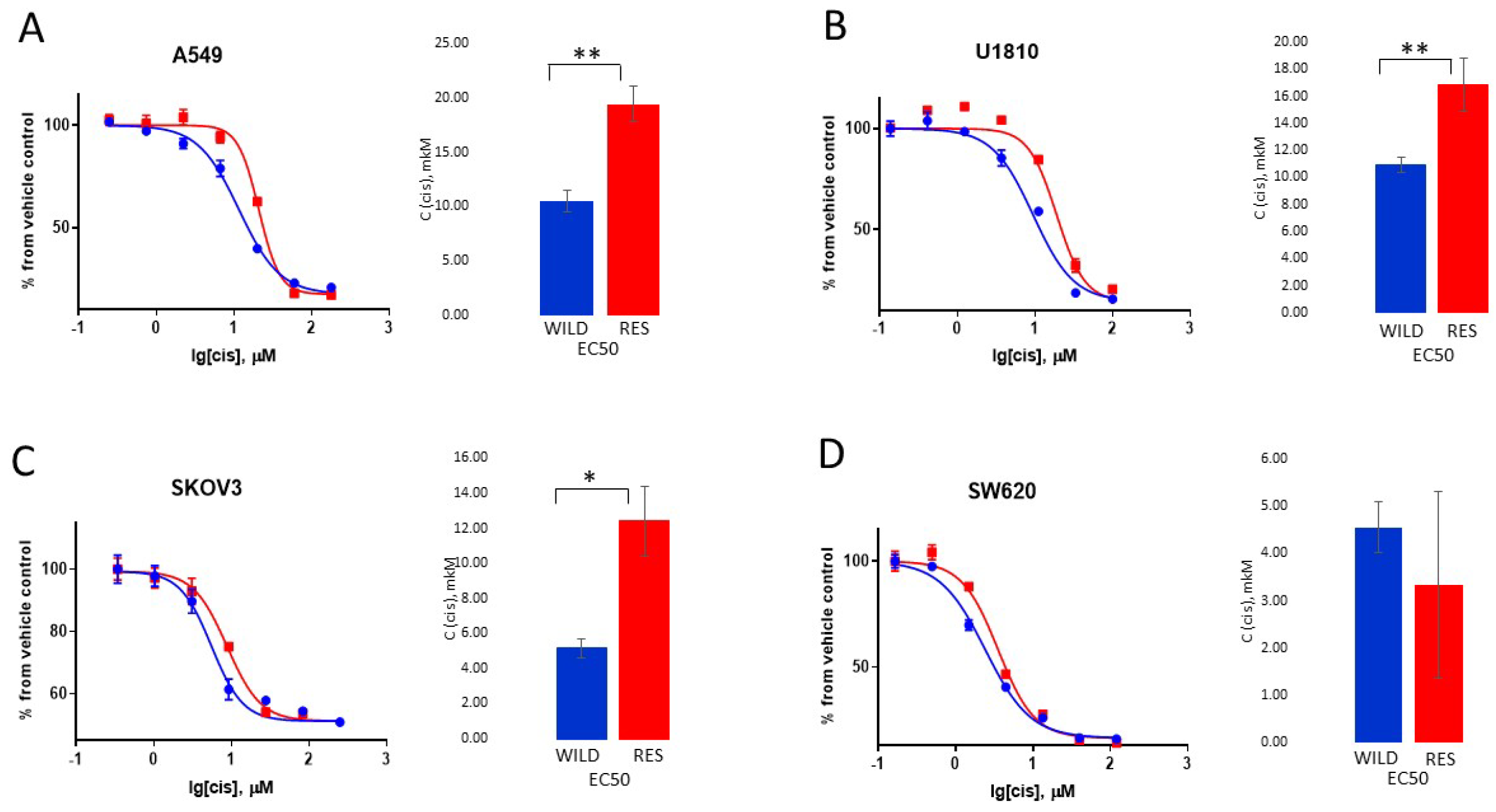
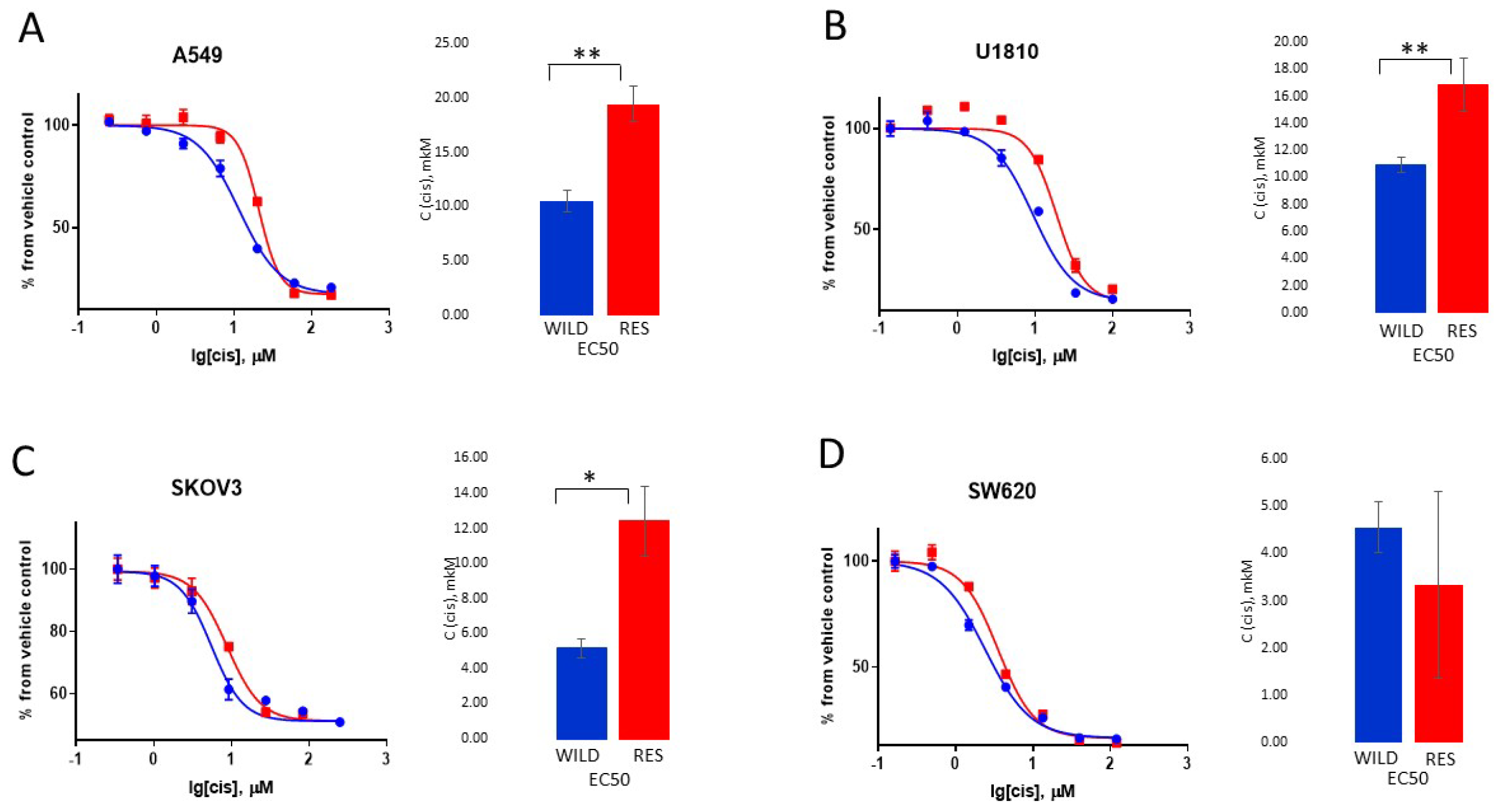
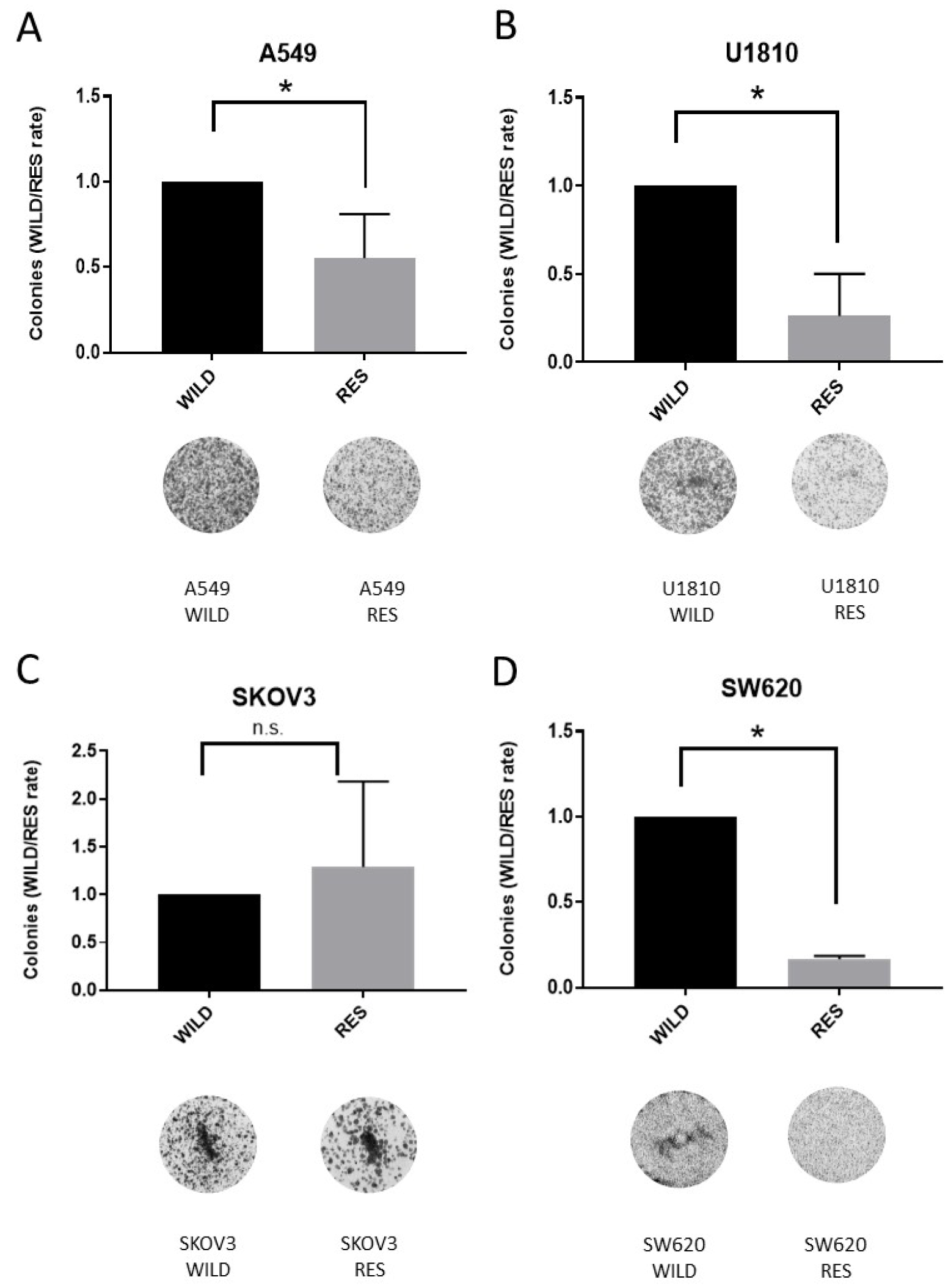
3.1. Creation and Characteristics of the Resistant Cell Lines
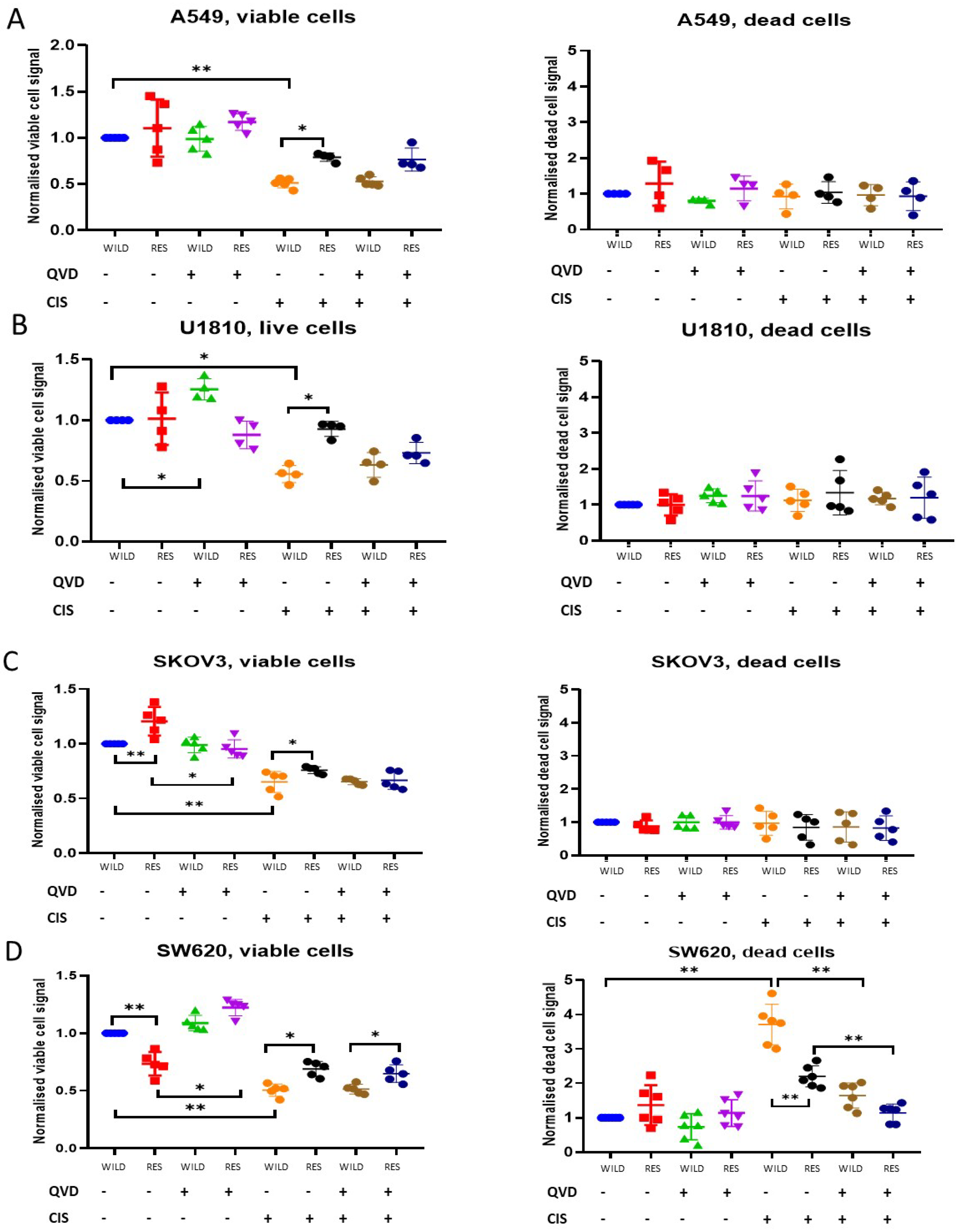
3.2. Cytotoxic and Cytostatic Effects of Cisplatin Treatment
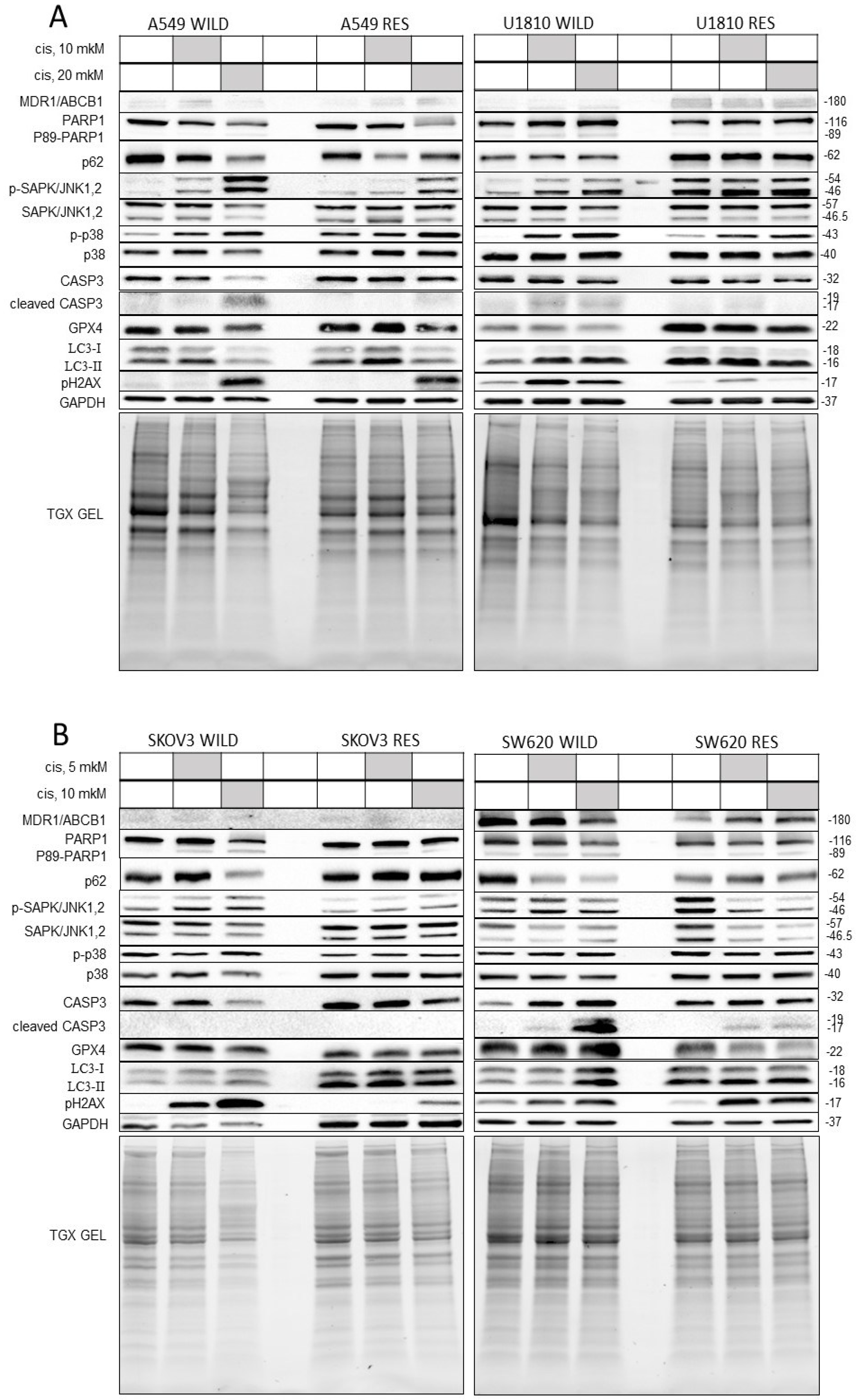
3.3. Biochemical Evaluation of Cisplatin Resistance
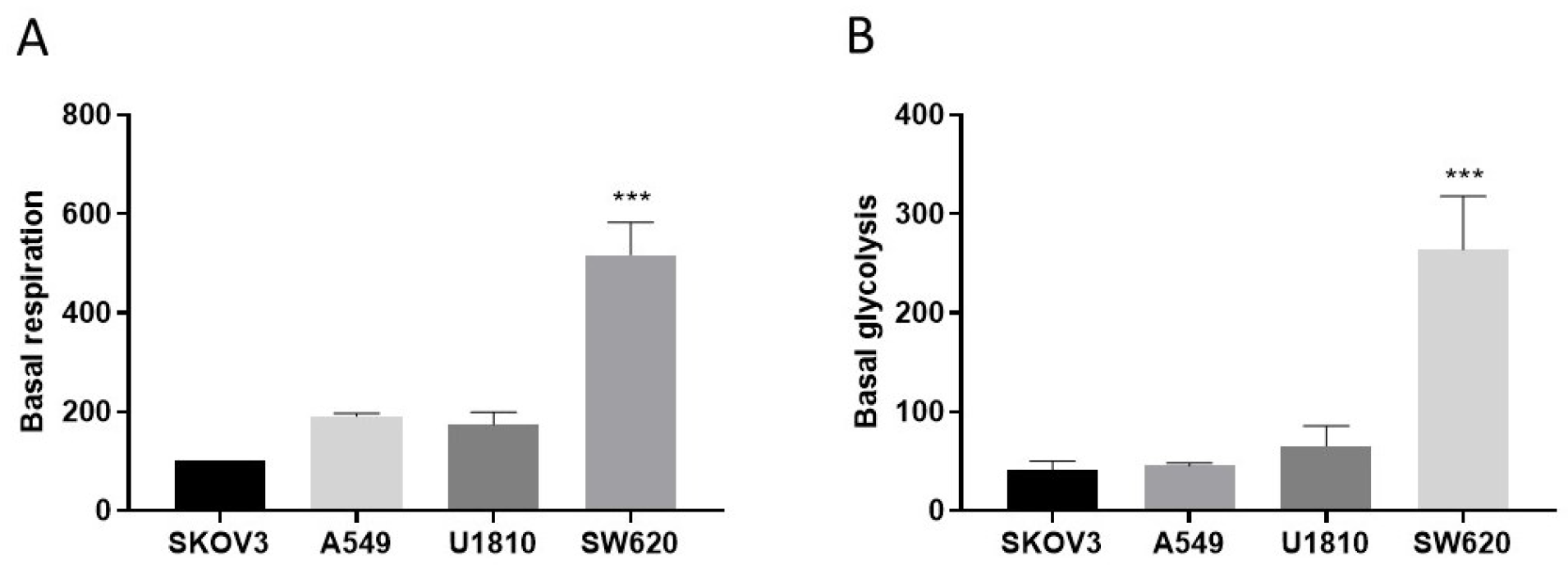
3.4. Metabolic Status in Cells Resistant to Cisplatin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Biological Hallmarks of Cancer. In Holland-Frei Cancer Medicine; Wiley: Hoboken, NJ, USA, 2017; pp. 1–10. [Google Scholar]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and Cancer—Role and Therapeutic Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]
- Bousset, L.; Gil, J. Targeting Senescence as an Anticancer Therapy. Mol. Oncol. 2022, 16, 3855–3880. [Google Scholar] [CrossRef]
- van Schaik, T.A.; Chen, K.-S.; Shah, K. Therapy-Induced Tumor Cell Death: Friend or Foe of Immunotherapy? Front. Oncol. 2021, 11, 678562. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated Cell Death (RCD) in Cancer: Key Pathways and Targeted Therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting Cell Death Pathways for Cancer Therapy: Recent Developments in Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis Research. J. Hematol. Oncol. 2022, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.V.S.; De Sousa Portilho, A.J.; Da Silva, E.L.; De Oliveira Sales, L.; Da Silva Maués, J.H.; De Moraes, M.E.A.; Moreira-Nunes, C.A. Establishment of Drug-Resistant Cell Lines as a Model in Experimental Oncology: A Review. Anticancer Res 2019, 39, 6443–6455. [Google Scholar] [CrossRef] [PubMed]
- Rosa, R.; Monteleone, F.; Zambrano, N.; Bianco, R. In Vitro and In Vivo Models for Analysis of Resistance to Anticancer Molecular Therapies. Curr. Med. Chem. 2014, 21, 1595–1606. [Google Scholar] [CrossRef]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; Oâ€TMDonovan, N.; Stordal, B. In Vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- Tippett, V.L.; Tattersall, L.; Ab Latif, N.B.; Shah, K.M.; Lawson, M.A.; Gartland, A. The Strategy and Clinical Relevance of in Vitro Models of MAP Resistance in Osteosarcoma: A Systematic Review. Oncogene 2023, 42, 259–277. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High Resistance to Cisplatin in Human Ovarian Cancer Cell Lines Is Associated with Marked Increase of Glutathione Synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef]
- Liu, W.-J.; Pan, P.-Y.; Sun, Y.; Wang, J.-B.; Zhou, H.; Xie, X.; Duan, Z.-Y.; Dong, H.-Y.; Chen, W.-N.; Zhang, L.; et al. Deferoxamine Counteracts Cisplatin Resistance in A549 Lung Adenocarcinoma Cells by Increasing Vulnerability to Glutamine Deprivation-Induced Cell Death. Front. Oncol. 2021, 11, 794735. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, G.; Condello, S.; Huang, H.; Cardenas, H.; Tanner, E.J.; Wei, J.; Ji, Y.; Li, J.; Tan, Y.; et al. Frizzled-7 Identifies Platinum-Tolerant Ovarian Cancer Cells Susceptible to Ferroptosis. Cancer Res. 2021, 81, 384–399. [Google Scholar] [CrossRef]
- Shenoy, P.; Chakraborty, S.; Gaikwad, S.; Sakpal, A.; Ray, P. Developing Clinically Relevant Acquired Chemoresistance Models in Epithelial Ovarian Cancer Cell Lines. Bio-Protocol 2022, 12, e4310. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.-Y.; Kim, H.-S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Chesnokov, M.S.; Zhivotovsky, B.; Kopeina, G.S. Drug Toxicity Assessment: Cell Proliferation versus Cell Death. Cell Death Discov. 2022, 8, 417. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, K.; Ambrose, A.R.; Davis, D.M. Escaping Death: How Cancer Cells and Infected Cells Resist Cell-Mediated Cytotoxicity. Front. Immunol. 2022, 13, 867098. [Google Scholar] [CrossRef] [PubMed]
- Fernald, K.; Kurokawa, M. Evading Apoptosis in Cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Werner, E. How Cancer Cells Escape Death. In Cellular Respiration and Carcinogenesis; Humana Press: Totowa, NJ, USA, 2009; pp. 161–178. [Google Scholar]
- Wang, Y.; Lina, L.; Xu, L.; Yang, Z.; Qian, Z.; Zhou, J.; Suoni, L. Arctigenin Enhances the Sensitivity of Cisplatin Resistant Colorectal Cancer Cell by Activating Autophagy. Biochem. Biophys. Res. Commun. 2019, 520, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Villeneuve, D.J.; Hembruff, S.L.; Kirwan, A.F.; Blais, D.E.; Bonin, M.; Parissenti, A.M. Cross-Resistance Studies of Isogenic Drug-Resistant Breast Tumor Cell Lines Support Recent Clinical Evidence Suggesting That Sensitivity to Paclitaxel May Be Strongly Compromised by Prior Doxorubicin Exposure. Breast Cancer Res. Treat. 2004, 85, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Jian, B.; Pang, J.; Xiong, H.; Zhang, W.; Zhan, T.; Su, Z.; Lin, H.; Zhang, H.; He, W.; Zheng, Y. Autophagy-Dependent Ferroptosis Contributes to Cisplatin-Induced Hearing Loss. Toxicol. Lett. 2021, 350, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Zamaraev, A.V.; Kopeina, G.S.; Buchbinder, J.H.; Zhivotovsky, B.; Lavrik, I.N. Caspase-2 Is a Negative Regulator of Necroptosis. Int. J. Biochem. Cell Biol. 2018, 102, 101–108. [Google Scholar] [CrossRef]
- Prokhorova, E.A.; Kopeina, G.S.; Lavrik, I.N.; Zhivotovsky, B. Apoptosis Regulation by Subcellular Relocation of Caspases. Sci. Rep. 2018, 8, 12199. [Google Scholar] [CrossRef]
- Sazonova, E.V.; Kopeina, G.S.; Imyanitov, E.N.; Zhivotovsky, B. Platinum Drugs and Taxanes: Can We Overcome Resistance? Cell Death Discov. 2021, 7, 155. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Furuya, K.N.; Bradley, G.; Sun, D.; Schuetz, E.G.; Schuetz, J.D. Identification of a New P-Glycoprotein-like ATP-Binding Cassette Transporter Gene That Is Overexpressed during Hepatocarcinogenesis. Cancer Res. 1997, 57, 3708–3716. [Google Scholar] [PubMed]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very Important Pharmacogene Summary. Pharmacogenet. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Ofir, R.; Seidman, R.; Rabinski, T.; Krup, M.; Yavelsky, V.; Weinstein, Y.; Wolfson, M. Taxol-Induced Apoptosis in Human SKOV3 Ovarian and MCF7 Breast Carcinoma Cells Is Caspase-3 and Caspase-9 Independent. Cell Death Differ. 2002, 9, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-KDa PARP1 Cleavage Fragment Serves as a Cytoplasmic PAR Carrier to Induce AIF-Mediated Apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef] [PubMed]
- Desroches, A.; Denault, J.-B. Caspase-7 Uses RNA to Enhance Proteolysis of Poly(ADP-Ribose) Polymerase 1 and Other RNA-Binding Proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 21521–21528. [Google Scholar] [CrossRef]
- Erener, S.; Pétrilli, V.; Kassner, I.; Minotti, R.; Castillo, R.; Santoro, R.; Hassa, P.O.; Tschopp, J.; Hottiger, M.O. Inflammasome-Activated Caspase 7 Cleaves PARP1 to Enhance the Expression of a Subset of NF-ΚB Target Genes. Mol. Cell 2012, 46, 200–211. [Google Scholar] [CrossRef]
- Dong, Y.; Xiong, M.; Duan, L.; Liu, Z.; Niu, T.; Luo, Y.; Wu, X.; Xu, C.; Lu, C. H2AX Phosphorylation Regulated by P38 Is Involved in Bim Expression and Apoptosis in Chronic Myelogenous Leukemia Cells Induced by Imatinib. Apoptosis 2014, 19, 1281–1292. [Google Scholar] [CrossRef]
- Sluss, H.K.; Davis, R.J. H2AX Is a Target of the JNK Signaling Pathway That Is Required For Apoptotic DNA Fragmentation. Mol. Cell 2006, 23, 152–153. [Google Scholar] [CrossRef]
- de la Cruz-Morcillo, M.A.; Valero, M.L.L.; Callejas-Valera, J.L.; Arias-González, L.; Melgar-Rojas, P.; Galán-Moya, E.M.; García-Gil, E.; García-Cano, J.; Sánchez-Prieto, R. P38MAPK Is a Major Determinant of the Balance between Apoptosis and Autophagy Triggered by 5-Fluorouracil: Implication in Resistance. Oncogene 2012, 31, 1073–1085. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, J.; Hou, H.; Li, J.; Li, J.; Liang, J.; Li, J.; Niu, X.; Hou, R.; Zhang, K. Autophagy Inhibits Inflammation via Down-Regulation of P38 MAPK/MTOR Signaling Cascade in Endothelial Cells. Clin. Cosmet. Investig. Dermatol. 2023, 16, 659–669. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yi, B.; Yang, S.; Liu, J.; Hu, J.; Wang, J.; Cao, K.; Zhang, W. VDR Activation Attenuate Cisplatin Induced AKI by Inhibiting Ferroptosis. Cell Death Dis. 2020, 11, 73. [Google Scholar] [CrossRef]
- Ercolani, L.; Florence, B.; Denaro, M.; Alexander, M. Isolation and Complete Sequence of a Functional Human Glyceraldehyde-3-Phosphate Dehydrogenase Gene. J. Biol. Chem. 1988, 263, 15335–15341. [Google Scholar] [CrossRef]
- Tisdale, E.J. Glyceraldehyde-3-Phosphate Dehydrogenase Is Phosphorylated by Protein Kinase Cι/λ and Plays a Role in Microtubule Dynamics in the Early Secretory Pathway. J. Biol. Chem. 2002, 277, 3334–3341. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Dorn, P.; Liu, S.; Deng, H.; Hall, S.R.R.; Peng, R.-W.; Schmid, R.A.; Marti, T.M. Cisplatin-Resistant A549 Non-Small Cell Lung Cancer Cells Can Be Identified by Increased Mitochondrial Mass and Are Sensitive to Pemetrexed Treatment. Cancer Cell Int. 2019, 19, 317. [Google Scholar] [CrossRef] [PubMed]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Associated with an Abrogation of Cisplatin-Induced G2/M Cell Cycle Arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef]
- Han, Y.; You, J.; Han, Y.; Liu, Y.; Huang, M.; Lu, X.; Chen, J.; Zheng, Y. LINC00184 Promotes Ovarian Cancer Cells Proliferation and Cisplatin Resistance by Elevating CNTN1 Expression via Sponging MiR-1305. Onco. Targets. Ther. 2021, 14, 2711–2726. [Google Scholar] [CrossRef] [PubMed]
- Fernández de Mattos, S.; Villalonga, P.; Clardy, J.; Lam, E.W.-F. FOXO3a Mediates the Cytotoxic Effects of Cisplatin in Colon Cancer Cells. Mol. Cancer Ther. 2008, 7, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-Y.; Liu, G.-H.; Olsson, B.; Sun, X.-F. Upregulation of the Antiapoptotic Factor Livin Contributes to Cisplatin Resistance in Colon Cancer Cells. Tumor Biol. 2013, 34, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.M.K.; Bateman, E.H. Tumor Control versus Adverse Events with Targeted Anticancer Therapies. Nat. Rev. Clin. Oncol. 2012, 9, 98–109. [Google Scholar] [CrossRef]
- Barr, M.P.; Gray, S.G.; Hoffmann, A.C.; Hilger, R.A.; Thomale, J.; O’Flaherty, J.D.; Fennell, D.A.; Richard, D.; O’Leary, J.J.; O’Byrne, K.J. Generation and Characterisation of Cisplatin-Resistant Non-Small Cell Lung Cancer Cell Lines Displaying a Stem-Like Signature. PLoS ONE 2013, 8, e54193. [Google Scholar] [CrossRef]
- Wang, D.; Ma, L.; Ma, Q.; Liu, J.; Jiang, G. Identification of New Biomarkers for Cisplatin-Resistance in A549 Human Lung Adenocarcinoma Cells by an Integrated Bioinformatical Analysis. Transl. Cancer Res. 2017, 6, 474–482. [Google Scholar] [CrossRef]
- Morelli, A.; Tortelli, T., Jr.; Pavan, I.; Silva, F.; Granato, D.; Peruca, G.; Pauletti, B.; Domingues, R.; Bezerra, R.; De Moura, L.; et al. Metformin Impairs Cisplatin Resistance Effects in A549 Lung Cancer Cells through MTOR Signaling and Other Metabolic Pathways. Int. J. Oncol. 2021, 58, 28. [Google Scholar] [CrossRef] [PubMed]
- Sergent, C.; Franco, N.; Chapusot, C.; Lizard-Nacol, S.; Isambert, N.; Correia, M.; Chauffert, B. Human Colon Cancer Cells Surviving High Doses of Cisplatin or Oxaliplatin in Vitro Are Not Defective in DNA Mismatch Repair Proteins. Cancer Chemother. Pharmacol. 2002, 49, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.A.; Kalinina, E.; Nuzhina, J.; Volodina, Y.; Shtil, A.; Tatarskiy, V. Potentiation of Cisplatin Cytotoxicity in Resistant Ovarian Cancer SKOV3/Cisplatin Cells by Quercetin Pre-Treatment. Int. J. Mol. Sci. 2023, 24, 10960. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-G.; Pak, J.H.; Choi, W.H.; Park, J.-Y.; Nam, J.-H.; Kim, J.-H. The Relationship between Cisplatin Resistance and Histone Deacetylase Isoform Overexpression in Epithelial Ovarian Cancer Cell Lines. J. Gynecol. Oncol. 2012, 23, 182. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sazonova, E.V.; Yapryntseva, M.A.; Pervushin, N.V.; Tsvetcov, R.I.; Zhivotovsky, B.; Kopeina, G.S. Cancer Drug Resistance: Targeting Proliferation or Programmed Cell Death. Cells 2024, 13, 388. https://doi.org/10.3390/cells13050388
Sazonova EV, Yapryntseva MA, Pervushin NV, Tsvetcov RI, Zhivotovsky B, Kopeina GS. Cancer Drug Resistance: Targeting Proliferation or Programmed Cell Death. Cells. 2024; 13(5):388. https://doi.org/10.3390/cells13050388
Chicago/Turabian StyleSazonova, Elena V., Maria A. Yapryntseva, Nikolay V. Pervushin, Roman I. Tsvetcov, Boris Zhivotovsky, and Gelina S. Kopeina. 2024. "Cancer Drug Resistance: Targeting Proliferation or Programmed Cell Death" Cells 13, no. 5: 388. https://doi.org/10.3390/cells13050388
APA StyleSazonova, E. V., Yapryntseva, M. A., Pervushin, N. V., Tsvetcov, R. I., Zhivotovsky, B., & Kopeina, G. S. (2024). Cancer Drug Resistance: Targeting Proliferation or Programmed Cell Death. Cells, 13(5), 388. https://doi.org/10.3390/cells13050388