

RL2 Enhances the Elimination of Breast Cancer Cells by Doxorubicin

 , and
, and 
Abstract
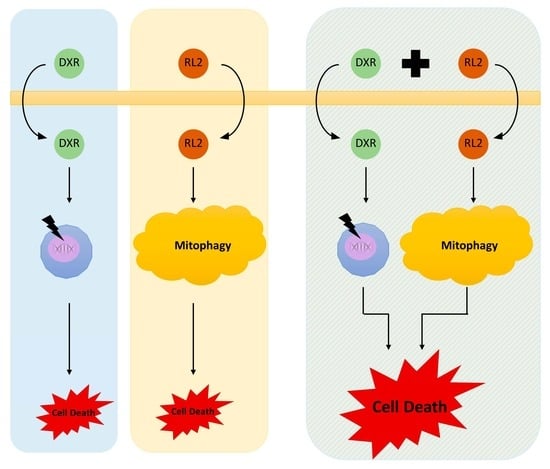
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Cell Viability Measurements by ATP and Metabolic (MT) Assays
2.4. Cytotoxicity Measurements by Lactatedehydrogenase (LDH) Assay
2.5. Caspase-3/7 Activity Assay
2.6. Western Blot Analysis
2.7. IC50 Value and CI
2.8. Statistics
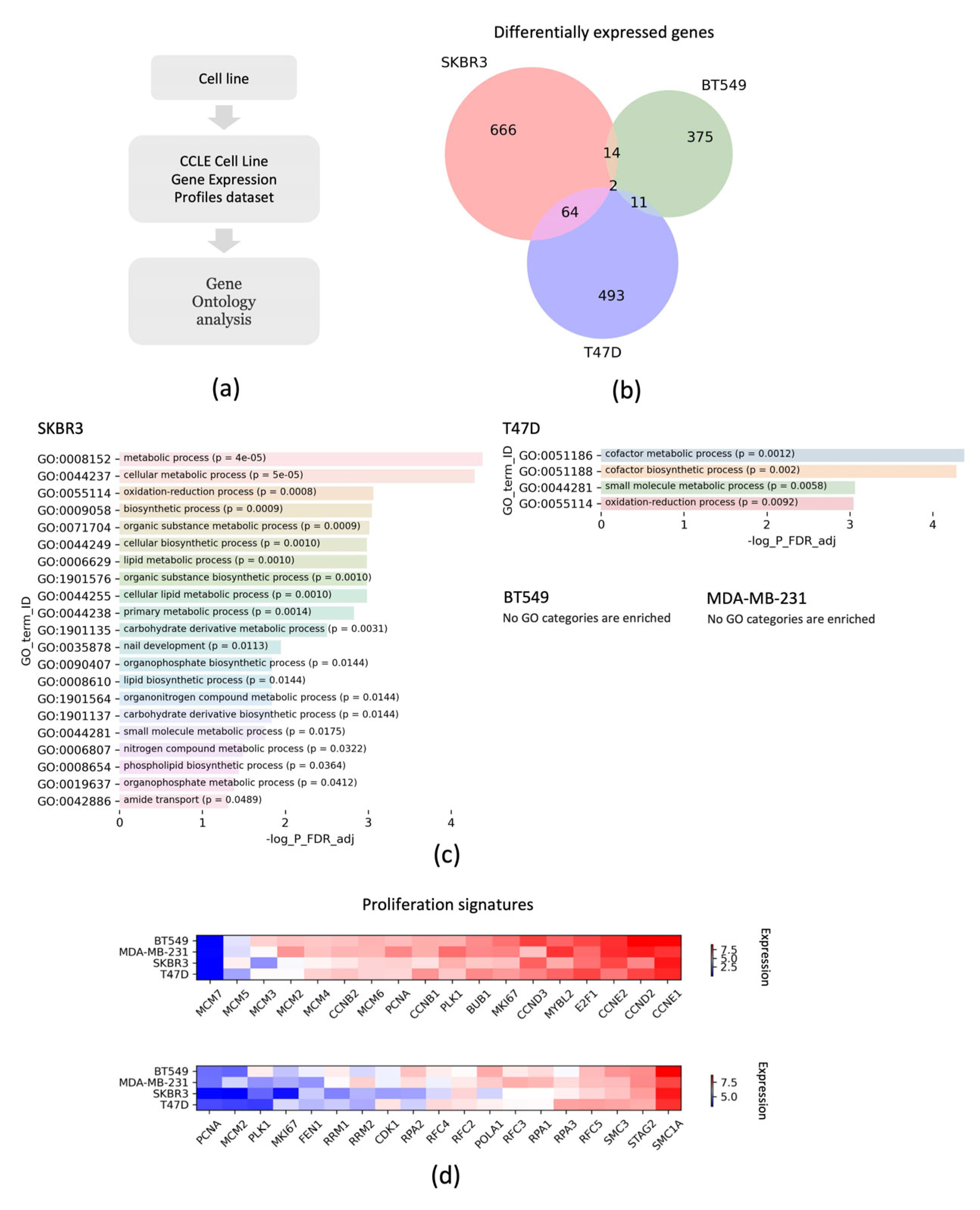
2.9. Bioinformatic Analysis
3. Results
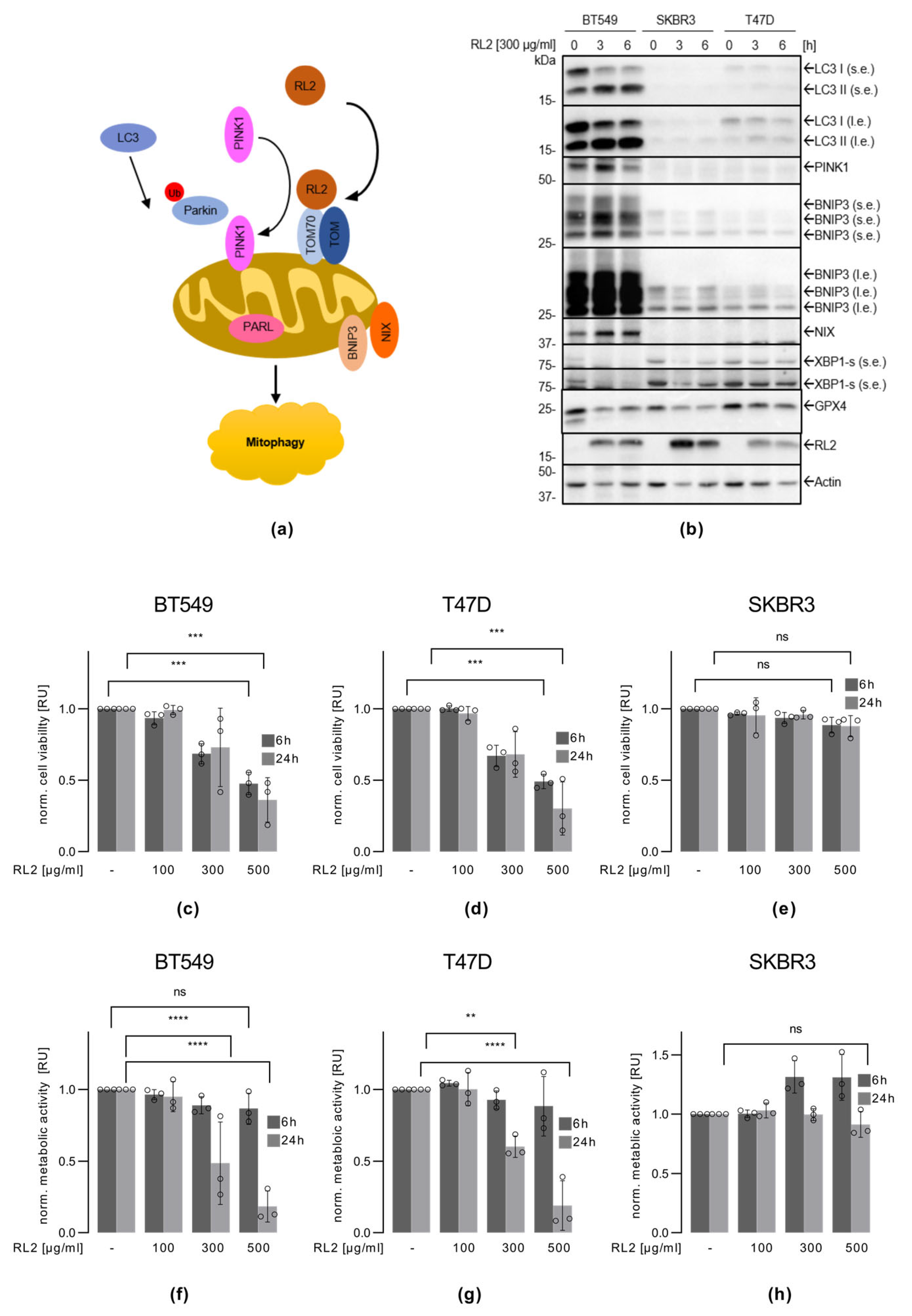
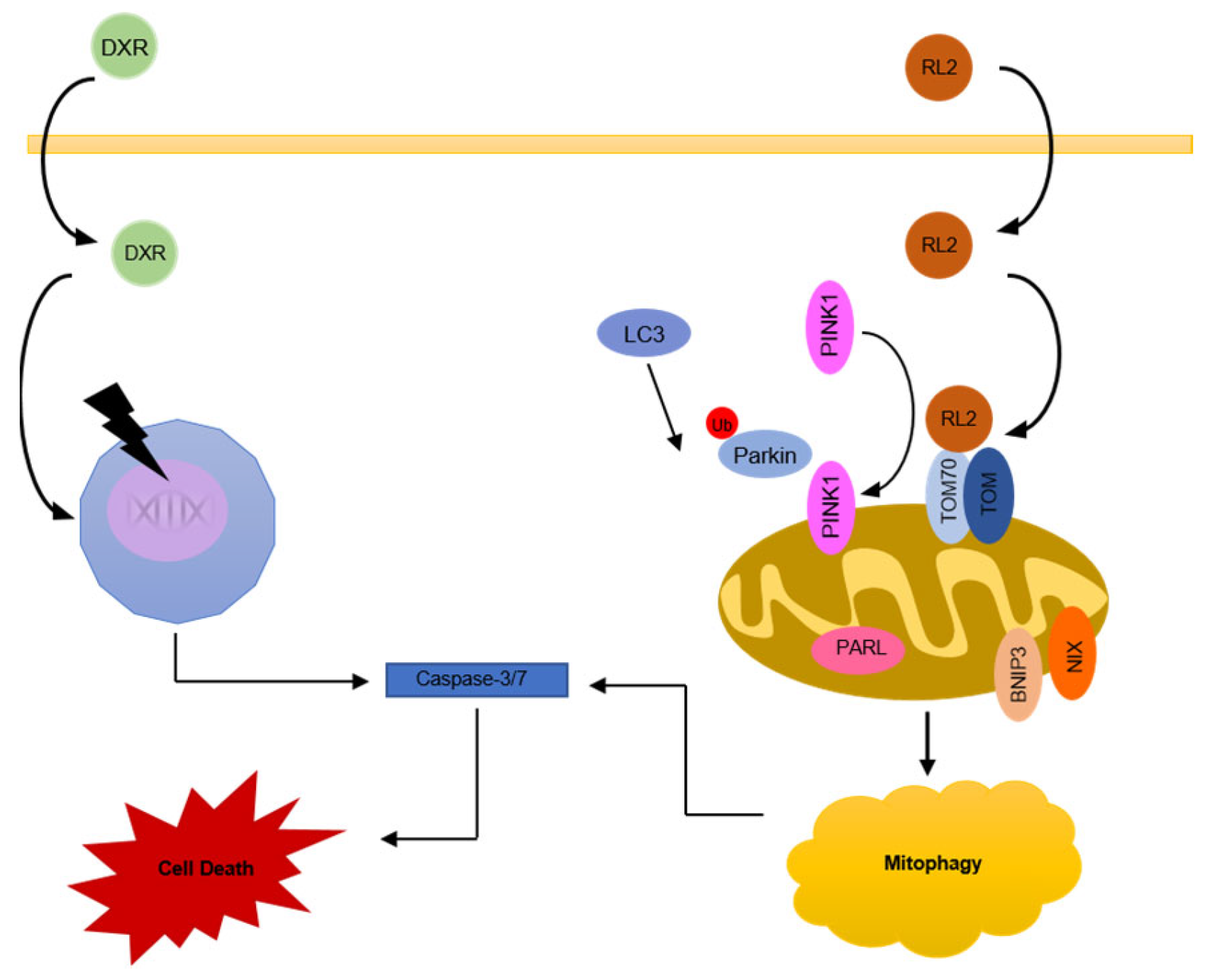
3.1. RL2 Induces Mitophagy in BT549 and T47D Breast Cancer Cells
3.2. RL2 Treatment Leads to the Viability Loss of BT549 and T47D Breast Cancer Cells
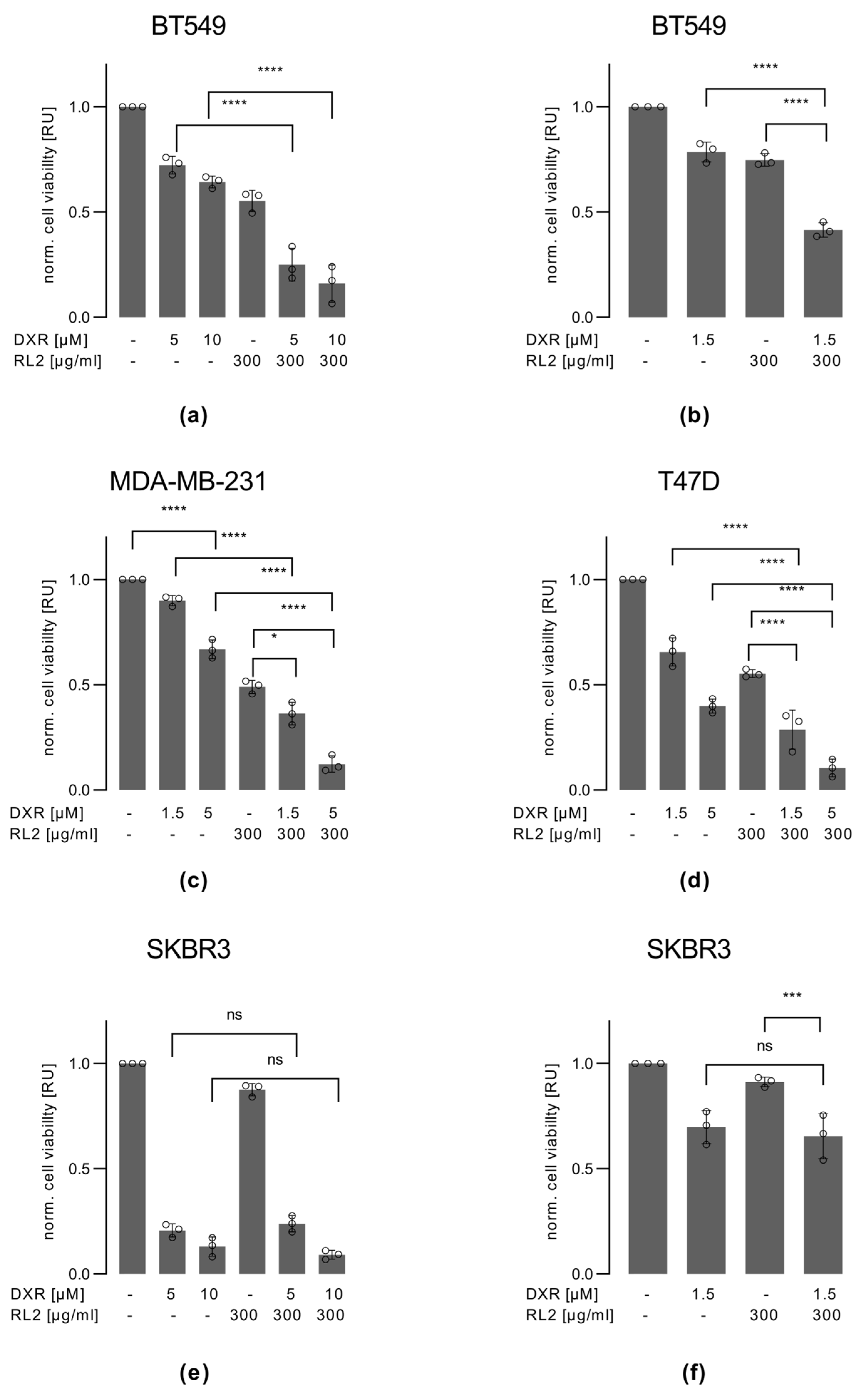
3.3. RL2 Enhances DXR-Induced Cell Viability Loss in BT549, MDA-MB-231 and T47D, but Not in SKBR3 Cells
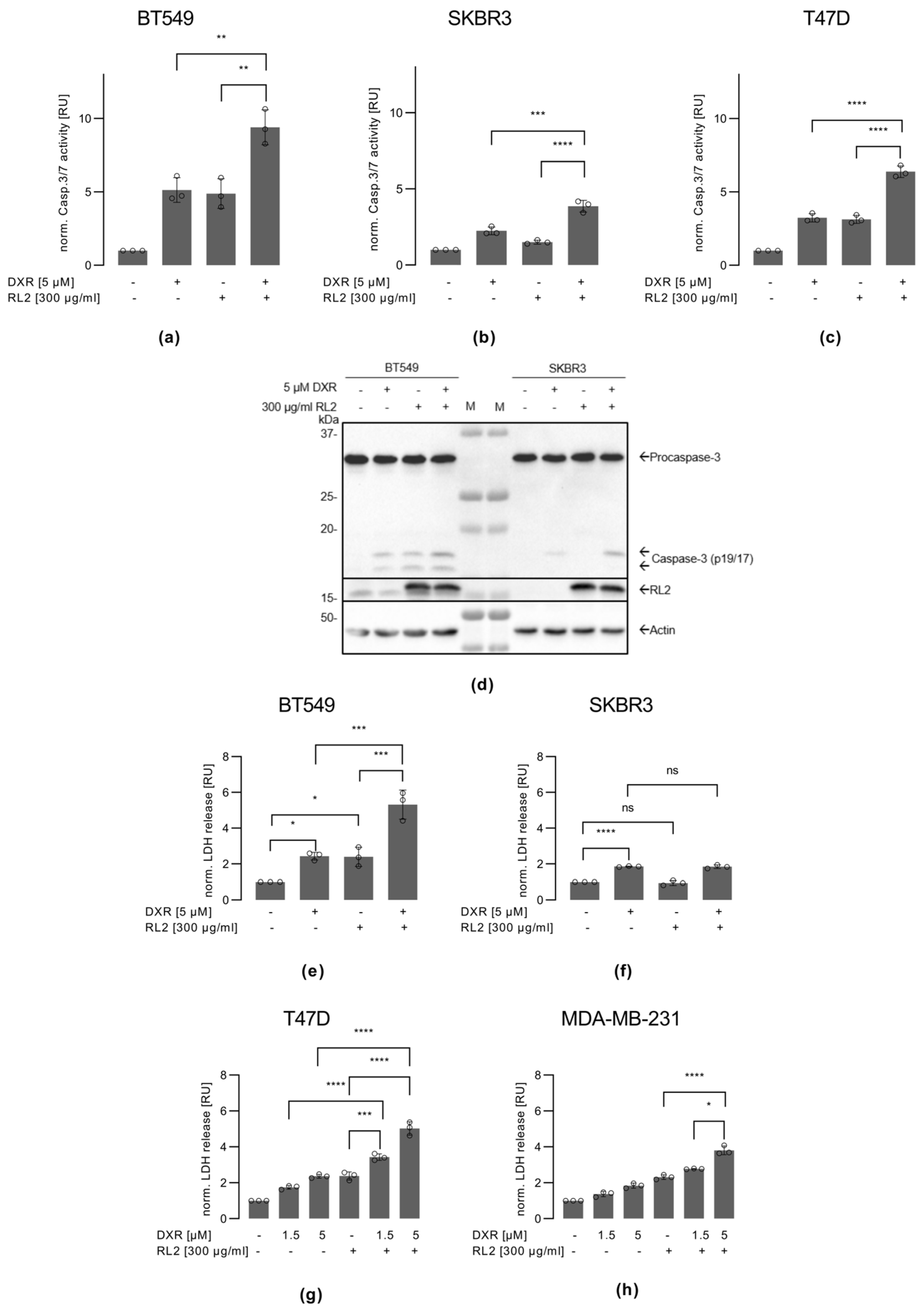
3.4. RL2 Enhances DXR-Induced Caspase-3 Activation and LDH Release
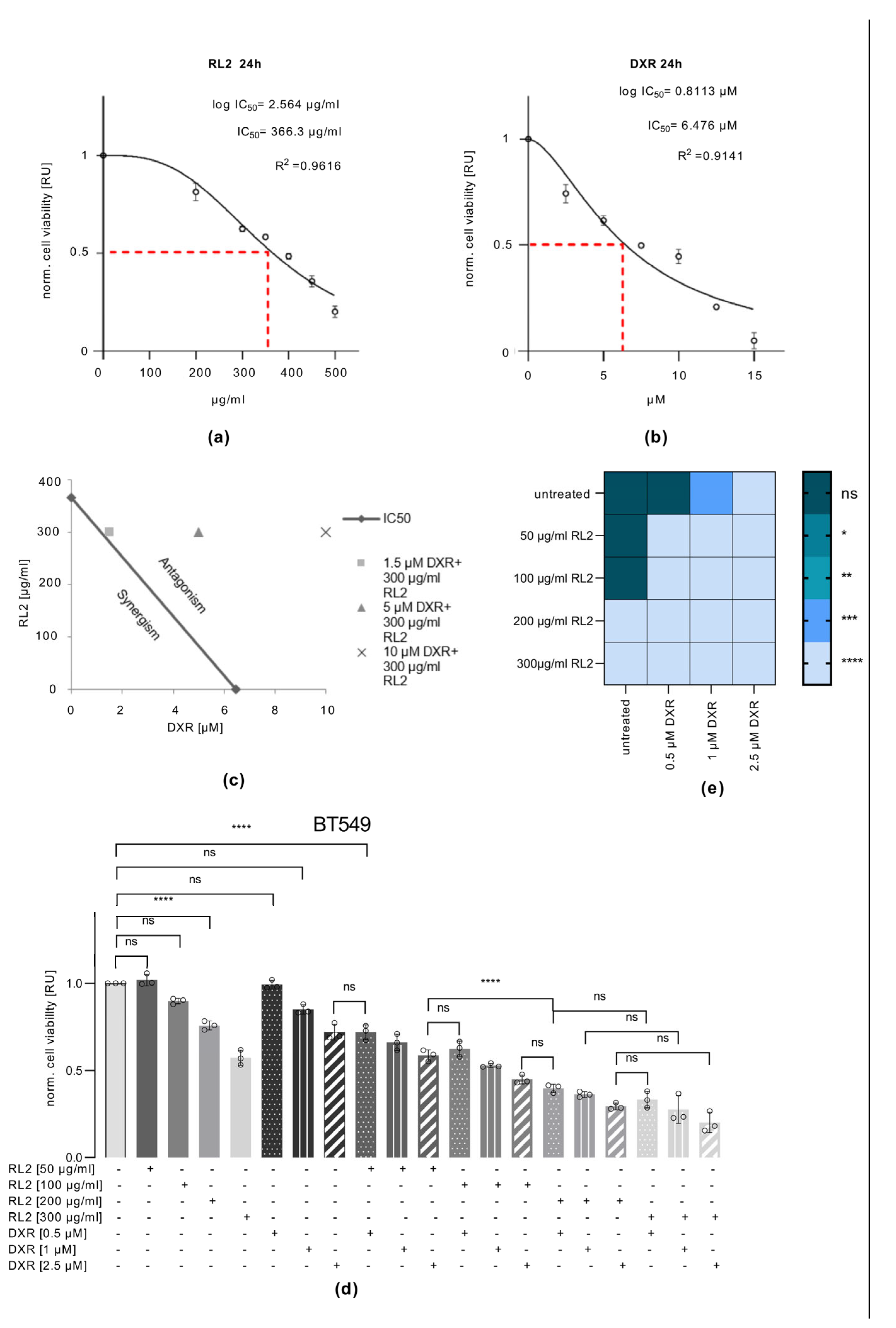
3.5. RL2 and DXR Have Synergistic Effects on Cell Viability Loss
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Seyrek, K.; Wohlfromm, F.; Espe, J.; Lavrik, I.N. The cross-talk of autophagy and apoptosis in breast carcinoma: Implications for novel therapies? Biochem. J. 2022, 479, 1581–1608. [Google Scholar] [CrossRef]
- Winters, S.; Martin, C.; Murphy, D.; Shikar, N.K. Breast Cancer Epidemiology, Prevention and Screening. Prog. Mol. Biol. Transl. Sci. 2017, 151, 1–32. [Google Scholar] [CrossRef]
- Bao, T.; Davidson, N.E. Gene Expression Profiling of Breast Cancer. Adv. Surg. 2008, 42, 249–260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferreira, M.A.; Gamazon, E.R.; Al-Ejeh, F.; Aittomäki, K.; Andrulis, I.L.; Anton-Culver, H.; Arason, A.; Arndt, V.; Aronson, K.J.; Arun, B.K.; et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nat. Commun. 2019, 10, 1741. [Google Scholar] [CrossRef] [PubMed]
- Ghislain, I.; Zikos, E.; Coens, C.; Quinten, C.; Balta, V.; Tryfonidis, K.; Piccart, M.; Zardavas, D.; Nagele, E.; Bjelic-Radisic, V.; et al. Health-related quality of life in locally advanced and metastatic breast cancer: Methodological and clinical issues in randomised controlled trials. Lancet Oncol. 2016, 17, e294–e304. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Thorburn, A. Targeting autophagy in breast cancer. World J. Clin. Oncol. 2014, 5, 224–240. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Fadillioǧlu, E.; Erdoǧan, H.; Söǧüt, S.; Kuku, I. Protective effects of erdosteine against doxorubicin-induced cardiomyopathy in rats. J. Appl. Toxicol. 2003, 23, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Pai, V.B.; Nahata, M.C. Cardiotoxicity of Chemotherapeutic Agents. Drug Saf. 2000, 22, 263–302. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, D.N.; Tardi, P.G.; Mayer, L.D.; Bally, M.B. A Comparison of Liposomal Formulations of Doxorubicin with Drug Administered in Free Form. Drug Saf. 2001, 24, 903–920. [Google Scholar] [CrossRef]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Semenov, D.V.; Fomin, A.S.; Kuligina, E.V.; Koval, O.A.; Matveeva, V.A.; Babkina, I.N.; Tikunova, N.V.; Richter, V.A. Recombinant analogs of a novel milk pro-apoptotic peptide, lactaptin, and their effect on cultured human cells. Protein J. 2010, 29, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kochneva, G.; Sivolobova, G.; Tkacheva, A.; Grazhdantseva, A.; Troitskaya, O.; Nushtaeva, A.; Tkachenko, A.; Kuligina, E.; Richter, V.; Koval, O. Engineering of double recombinant vaccinia virus with enhanced oncolytic potential for solid tumor virotherapy. Oncotarget 2016, 7, 74171–74188. [Google Scholar] [CrossRef] [PubMed]
- Koval, O.A.; Fomin, A.S.; Kaledin, V.I.; Semenov, D.V.; Potapenko, M.O.; Kuligina, E.V.; Nikolin, V.P.; Nikitenko, E.V.; Richter, V.A. A novel pro-apoptotic effector lactaptin inhibits tumor grwoth in mice models. Biochimie 2012, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Koval, O.A.; Tkachenko, A.V.; Fomin, A.S.; Semenov, D.V.; Nushtaeva, A.A.; Kuligina, E.V.; Zavjalov, E.L.; Richter, V.A. Lactaptin Induces p53-Independent Cell Death Associated with Features of Apoptosis and Autophagy and Delays Growth of Breast Cancer Cells in Mouse Xenografts. PLoS ONE 2014, 9, e93921. [Google Scholar] [CrossRef]
- Chinak, O.A.; Shernyukov, A.V.; Ovcherenko, S.S.; Sviridow, E.A.; Golyshev, V.M.; Fomin, A.S.; Pyshnaya, I.A.; Kuligina, E.V.; Richter, V.A.; Bagryanskaya, E.G. Structural and Aggregation Features of a Human K-Casein Fragment with Antitumor and Cell-Penetrating Properties. Molecules 2019, 24, 2919. [Google Scholar] [CrossRef]
- Richter, M.; Wohlfromm, F.; Kähne, T.; Bongartz, H.; Seyrek, H.; Kit, Y.; Chinak, O.; Richter, V.A.; Koval, O.A.; Lavrik, I.N. The Recombinant Fragment of Human K-Casein Induces cell Death by Targeting the Proteins of the Mitochondrial Import in Breast Cancer Cells. Cancers 2020, 12, 1427. [Google Scholar] [CrossRef]
- Bagamanshina, A.V.; Troitskaya, O.S.; Nushtaeva, A.A.; Yunusova, A.Y.; Starykovych, M.O.; Kuligina, E.V.; Kit, Y.Y.; Richter, M.; Wohlfromm, F.; Kähne, T.; et al. Cytotoxic and Antitumor Activity of Lactaptin in Combination with Autophagy Inducers and Inhibitors. BioMed Res. Int. 2019, 2019, 4087160. [Google Scholar] [CrossRef]
- Wohlfromm, F.; Richter, M.; Otrin, L.; Seyrek, K.; Vidaković-Koch, T.; Kuligina, E.; Richter, V.; Koval, O.; Lavrik, I.N. Interplay Between Mitophagy and Apoptosis Defines a Cell Fate Upon Co-treatment of Breast Cancer Cells With a Recombinant Fragment of Human κ-Casein and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand. Front. Cell Dev. Biol. 2021, 8, 617762. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Vale, N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100110. [Google Scholar] [CrossRef] [PubMed]
- Pomaznoy, M.; Ha, B.; Peters, B. GOnet: A tool for interactive Gene Ontology analysis. BMC Bioinform. 2018, 19, 470. [Google Scholar] [CrossRef] [PubMed]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R.; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e316. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database J. Biol. Databases Curation 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic Reticulum Stress Triggers Autophagy*. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Ueno, M.; Kakinuma, Y.; Yuhki, K.-i.; Murakoshi, N.; Iemitsu, M.; Miyauchi, T.; Yamaguchi, I. Doxorubicin Induces Apoptosis by Activation of Caspase-3 in Cultured Cardiomyocytes In Vitro and Rat Cardiac Ventricles In Vivo. J. Pharmacol. Sci. 2006, 101, 151–158. [Google Scholar] [CrossRef]
- Thayaparasingham, B.; Kunz, A.; Peters, N.; Kulms, D. Sensitization of melanoma cells to TRAIL by UVB-induced and NF-kappaB-mediated downregulation of xIAP. Oncogene 2009, 28, 345–362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hörnle, M.; Peters, N.; Thayaparasingham, B.; Vörsmann, H.; Kashkar, H.; Kulms, D. Caspase-3 cleaves XIAP in a positive feedback loop to sensitize melanoma cells to TRAIL-induced apoptosis. Oncogene 2011, 30, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Chinak, O.; Golubitskaya, E.; Pyshnaya, I.; Stepanov, G.; Zhuravlev, E.; Richter, V.; Koval, O. Nucleic Acids Delivery Into the Cells Using Pro-Apoptotic Protein Lactaptin. Front. Pharmacol. 2019, 10, 1043. [Google Scholar] [CrossRef] [PubMed]
- Ovcherenko, S.S.; Chinak, O.A.; Chechushkov, A.V.; Dobrynin, S.A.; Kirilyuk, I.A.; Krumkacheva, O.A.; Richter, V.A.; Bagryanskaya, E.G. Uptake of Cell-Penetrating Peptide RL2 by Human Lung Cancer Cells: Monitoring by Electron Paramagnetic Resonance and Confocal Laser Scanning Microscopy. Molecules 2021, 26, 5442. [Google Scholar] [CrossRef] [PubMed]
- Subrakova, V.G.; Kulemzin, S.V.; Belovezhets, T.N.; Chikaev, A.N.; Chikaev, N.A.; Koval, O.A.; Gorchakov, A.A.; Taranin, A.V. shp-2 gene knockout upregulates CAR-driven cytotoxicity of YT NK cells. Vavilovskii Zhurnal Genet. I Sel. 2020, 24, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Koval, O.A.; Subrakova, V.G.; Nushtaeva, A.A.; Belovezhets, T.N.; Troitskaya, O.A.; Ermakov, M.S.; Varlamov, M.E.; Chikaev, A.N.; Kuligina, E.V.; Kulemzin, S.V.; et al. CAR-dependent anti-metastatic activity of modified NK cell line YT. Genes Cells 2019, 14, 66–71. [Google Scholar] [CrossRef]
- Tkachenko, A.V.; Troitskaya, O.S.; Semenov, D.V.; Dmitrienko, E.V.; Kuligina, E.V.; Richter, V.A.; Koval, O.A. Immunogenicity of recombinant analog of antitumor protein lactaptin. Mol. Biol. 2017, 51, 687–694. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Xu, S.; Wang, S.; Tu, Y.; Xiong, Y.; Mei, J.; Wang, C. The role of MAPK signaling pathway in the Her-2-positive meningiomas. Oncol. Rep. 2016, 36, 685–695. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wohlfromm, F.; Seyrek, K.; Ivanisenko, N.; Troitskaya, O.; Kulms, D.; Richter, V.; Koval, O.; Lavrik, I.N. RL2 Enhances the Elimination of Breast Cancer Cells by Doxorubicin. Cells 2023, 12, 2779. https://doi.org/10.3390/cells12242779
Wohlfromm F, Seyrek K, Ivanisenko N, Troitskaya O, Kulms D, Richter V, Koval O, Lavrik IN. RL2 Enhances the Elimination of Breast Cancer Cells by Doxorubicin. Cells. 2023; 12(24):2779. https://doi.org/10.3390/cells12242779
Chicago/Turabian StyleWohlfromm, Fabian, Kamil Seyrek, Nikita Ivanisenko, Olga Troitskaya, Dagmar Kulms, Vladimir Richter, Olga Koval, and Inna N. Lavrik. 2023. "RL2 Enhances the Elimination of Breast Cancer Cells by Doxorubicin" Cells 12, no. 24: 2779. https://doi.org/10.3390/cells12242779
APA StyleWohlfromm, F., Seyrek, K., Ivanisenko, N., Troitskaya, O., Kulms, D., Richter, V., Koval, O., & Lavrik, I. N. (2023). RL2 Enhances the Elimination of Breast Cancer Cells by Doxorubicin. Cells, 12(24), 2779. https://doi.org/10.3390/cells12242779