
Single-Cell RNA Sequencing Reveals HIF1A as a Severity-Sensitive Immunological Scar in Circulating Monocytes of Convalescent Comorbidity-Free COVID-19 Patients


Abstract
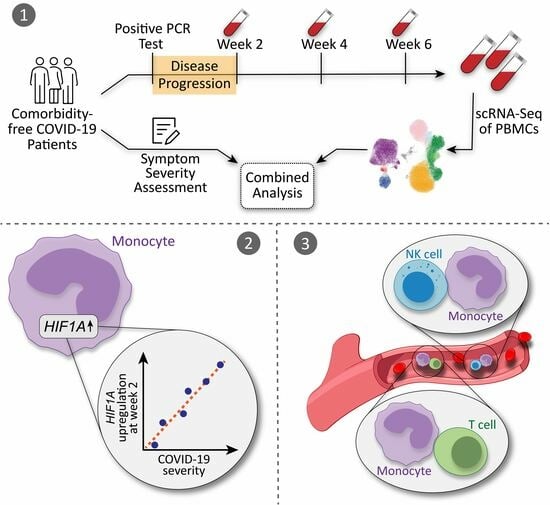
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
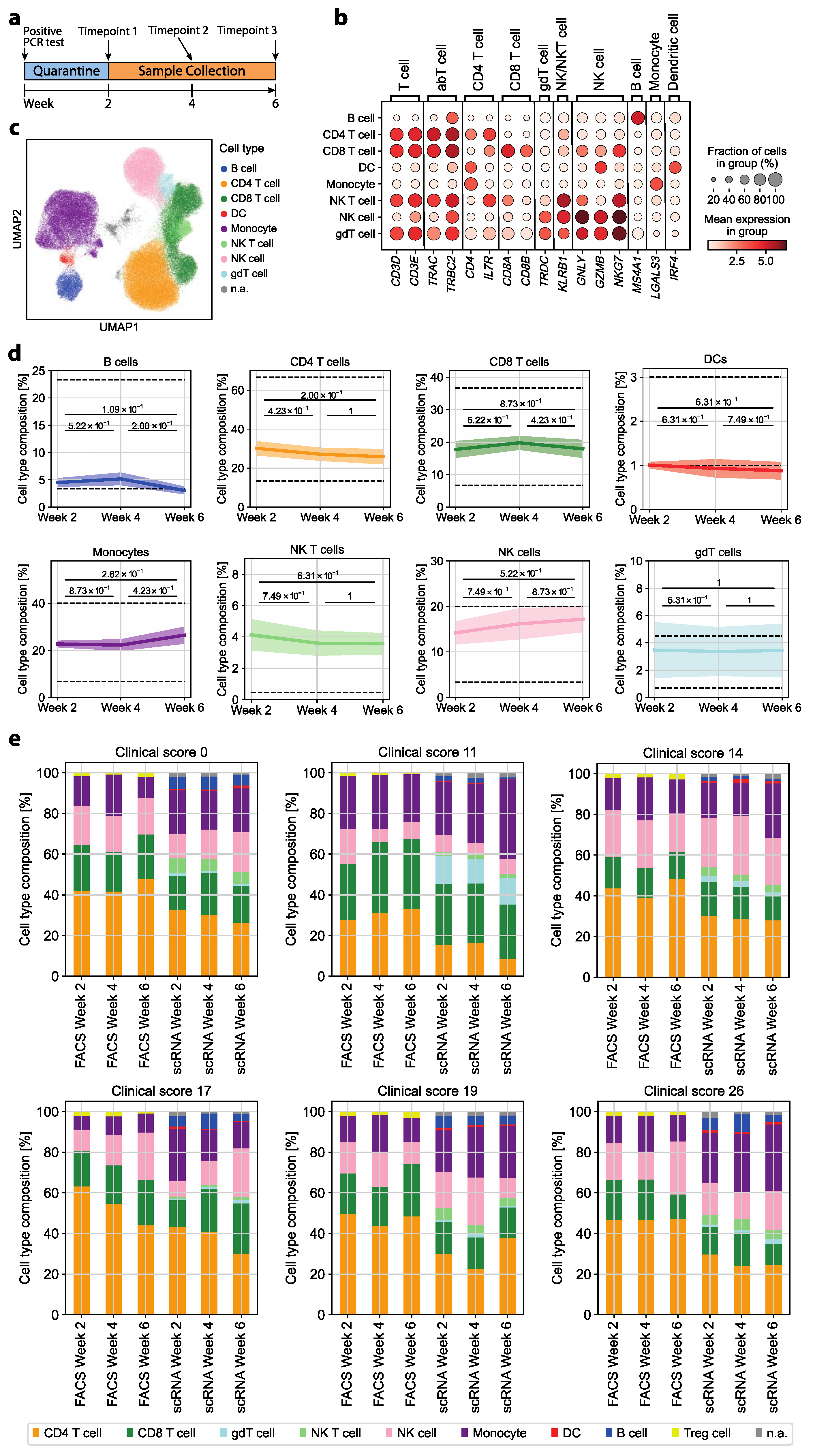
2.1. Patient Cohort Characterization
2.2. Cell Isolation and Single-Cell RNA Sequencing (scRNA-seq)
2.3. scRNA-seq Data Analysis
2.4. Statistics
3. Results
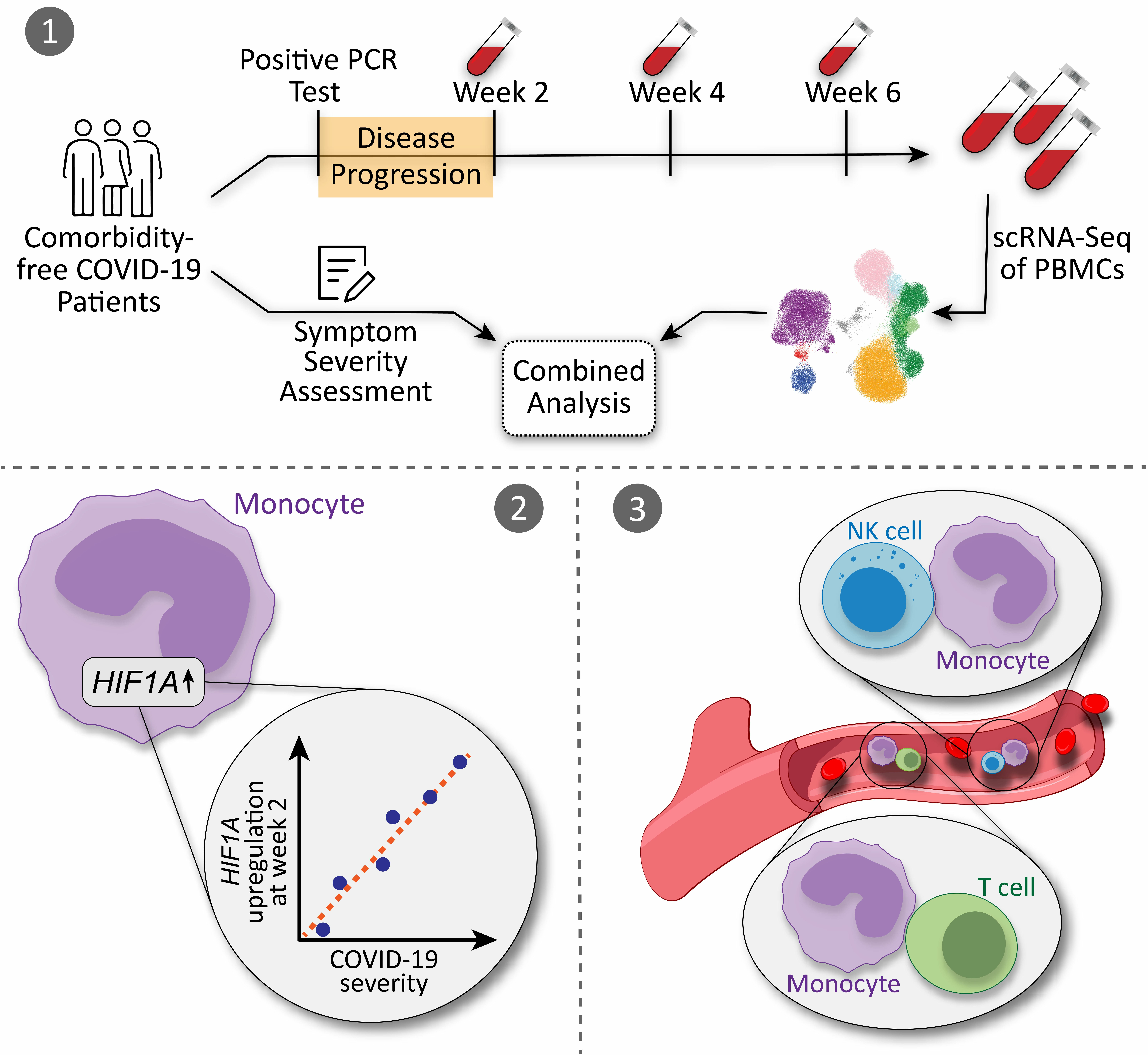
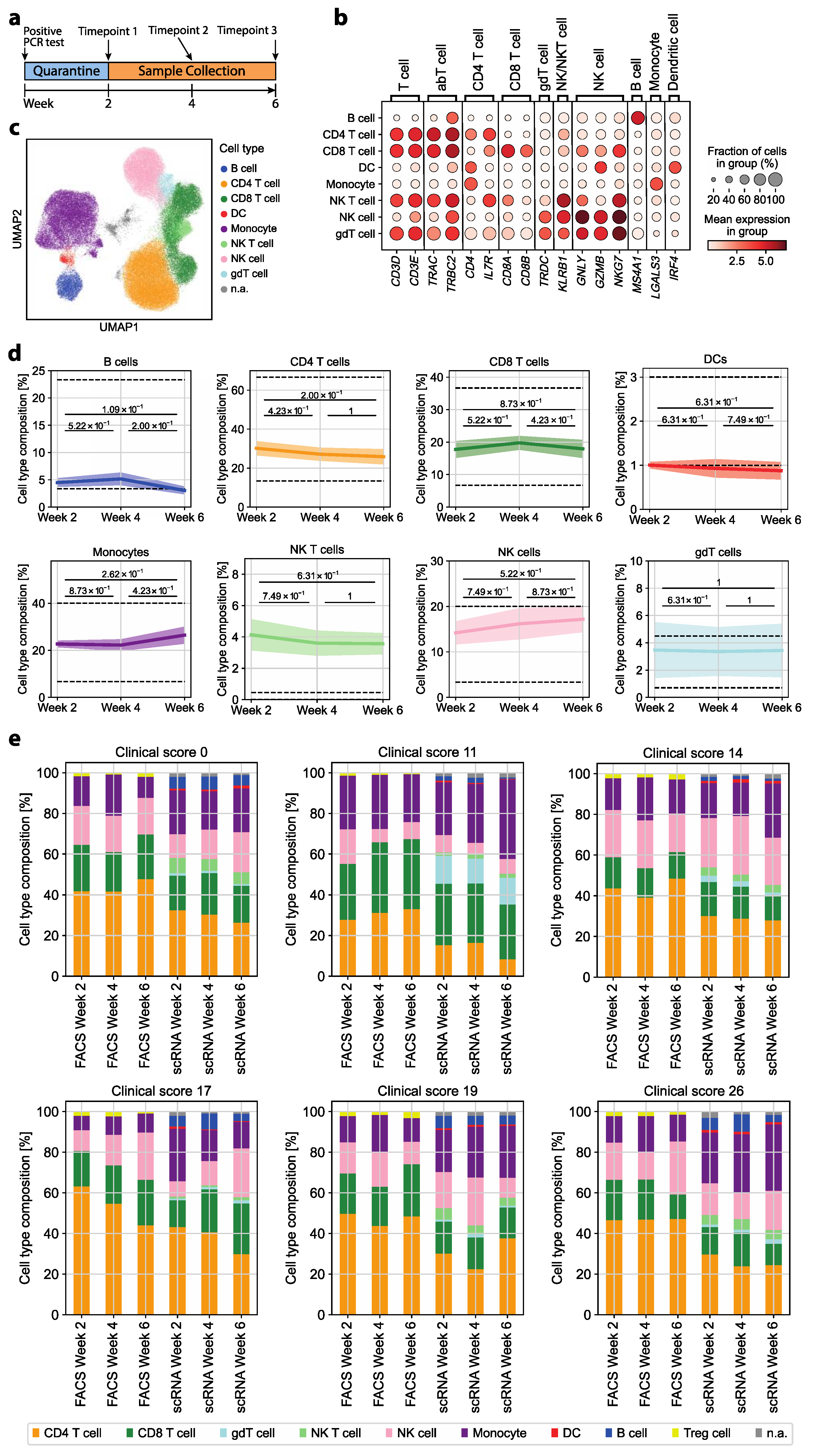
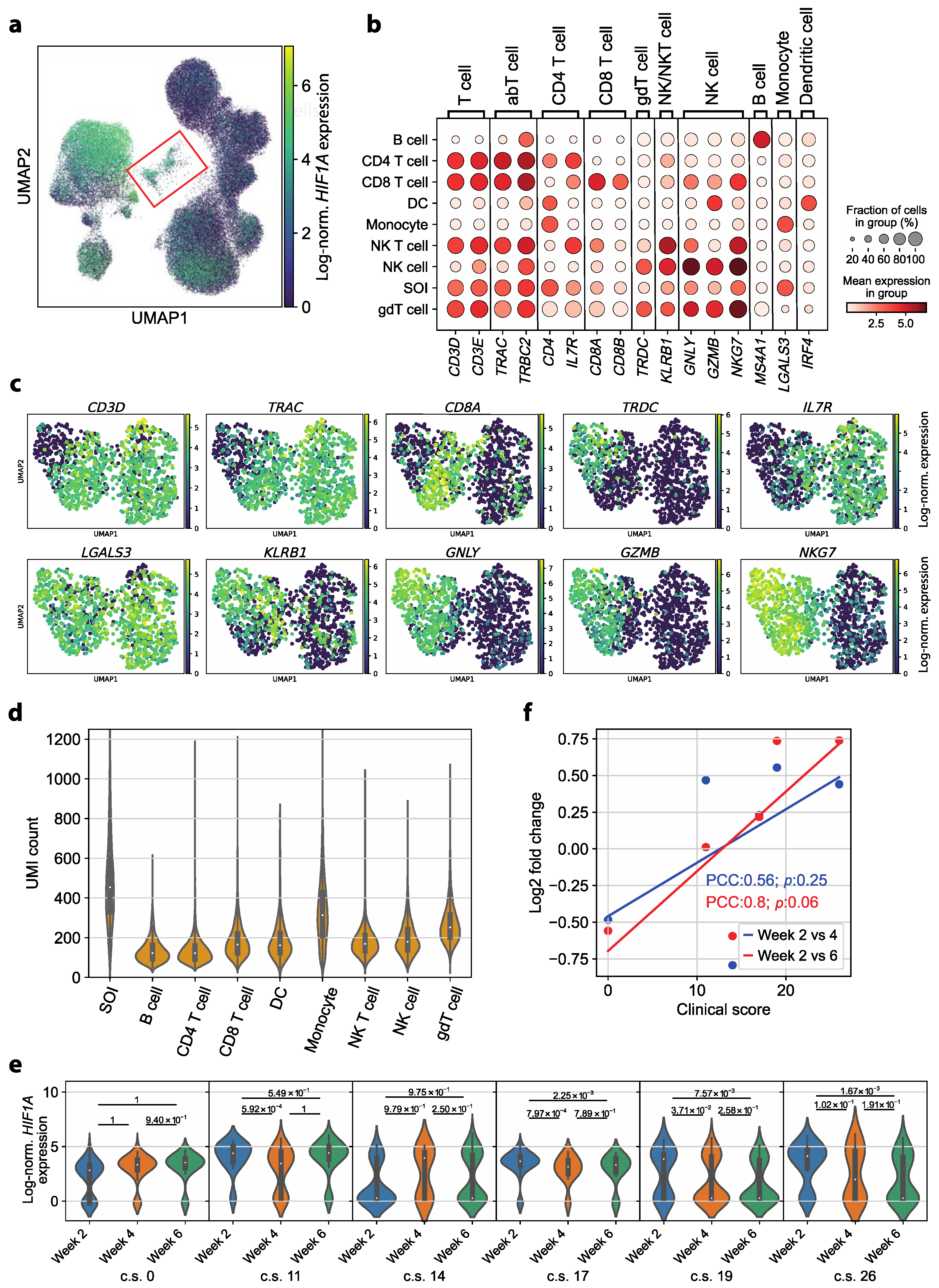
3.1. The Immune Cell Composition of Convalescent COVID-19 Patients Demonstrates Interindividual Stability over Time
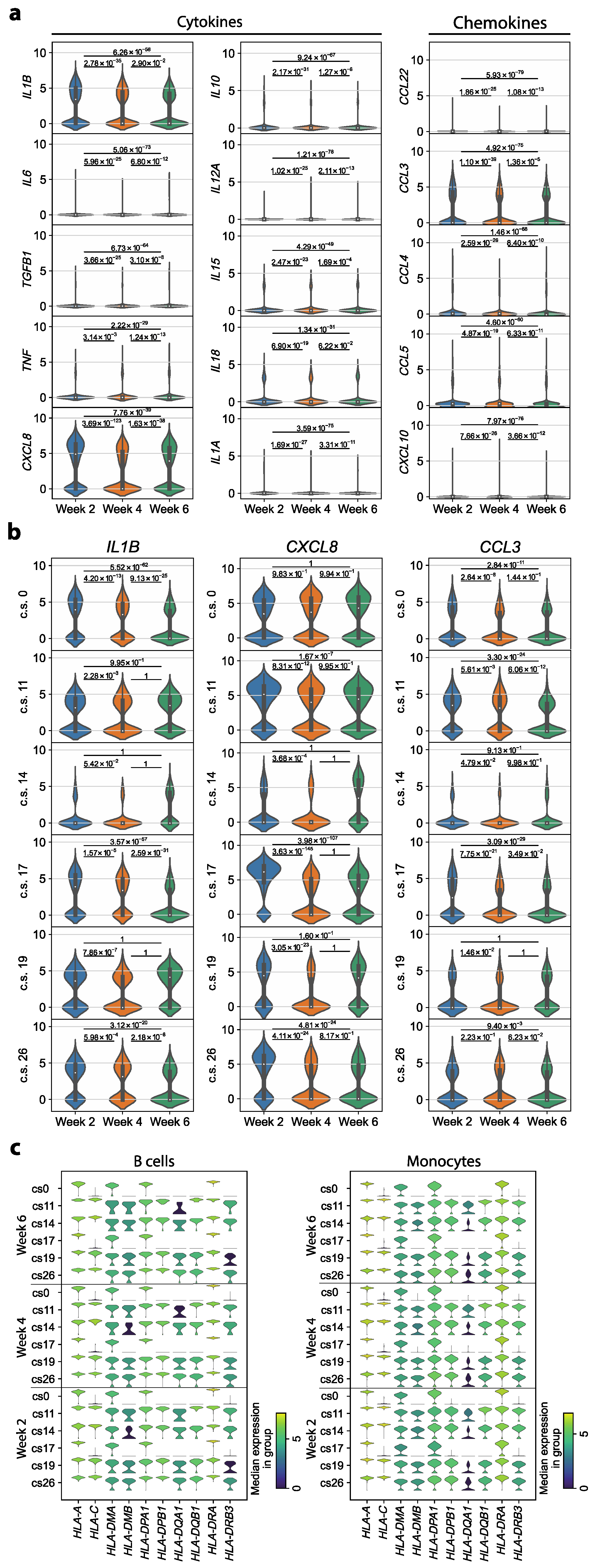
3.2. Transcriptomic Dysregulations Typical for COVID-19 Are Absent in Early Convalescent Patients
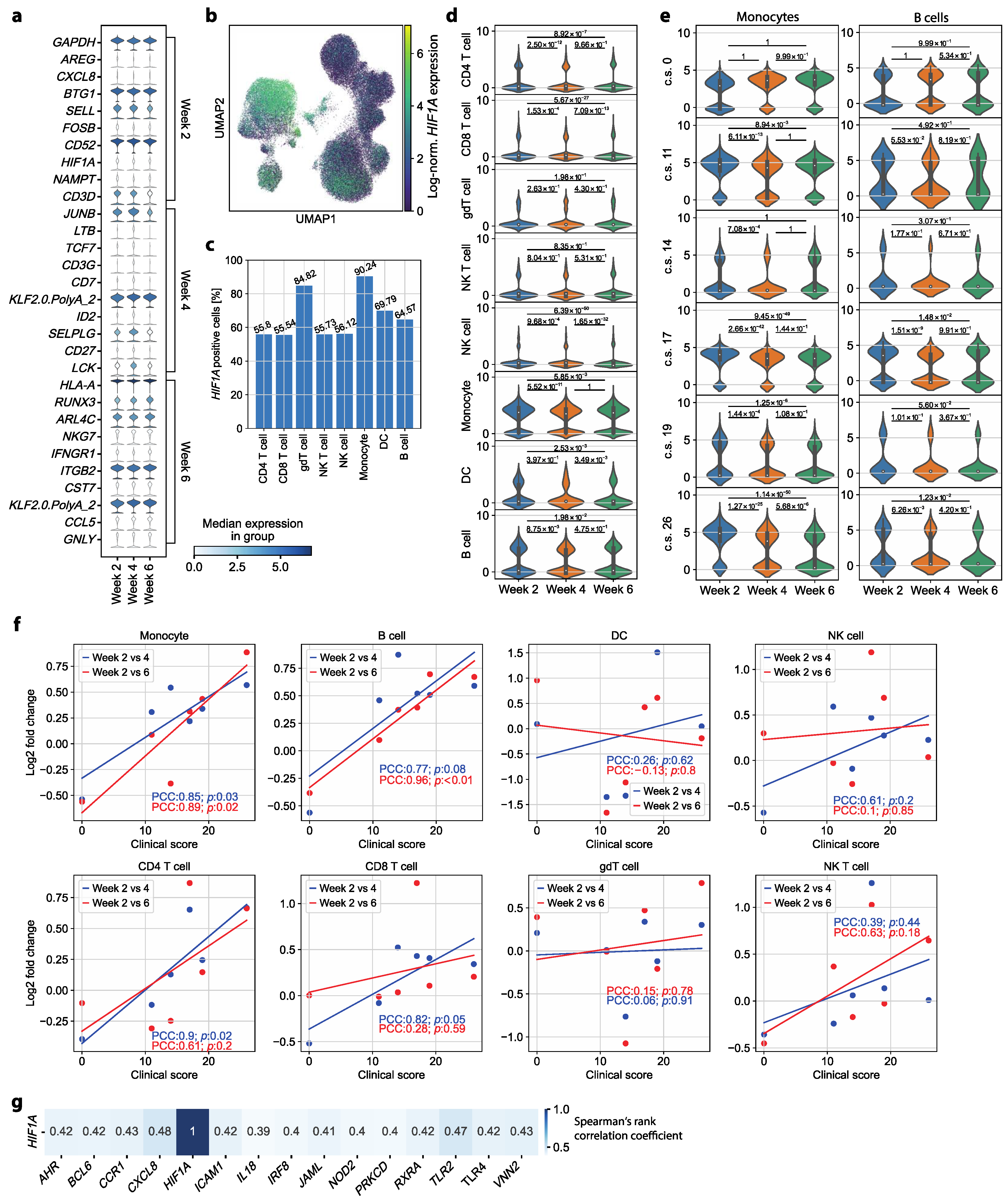
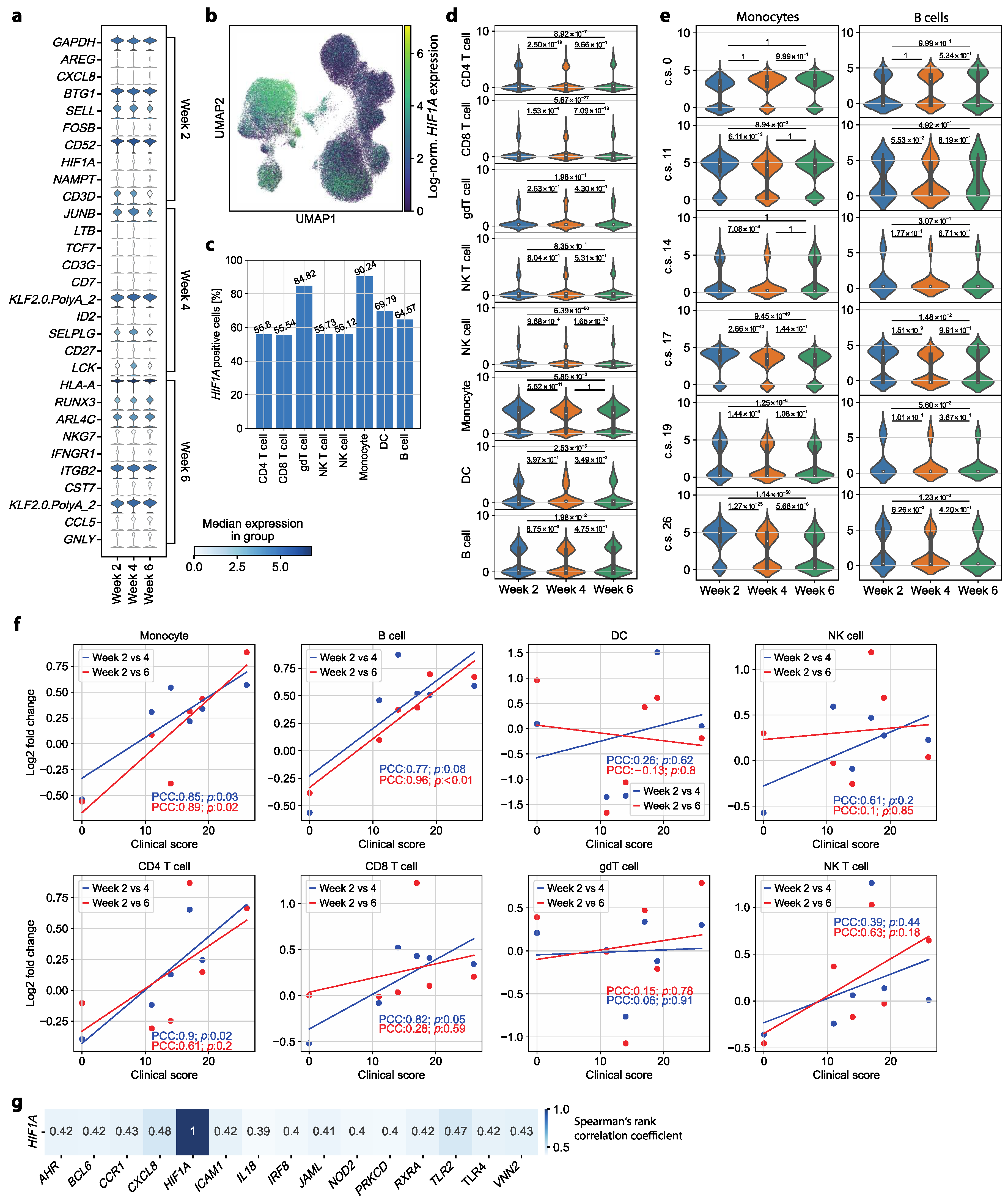
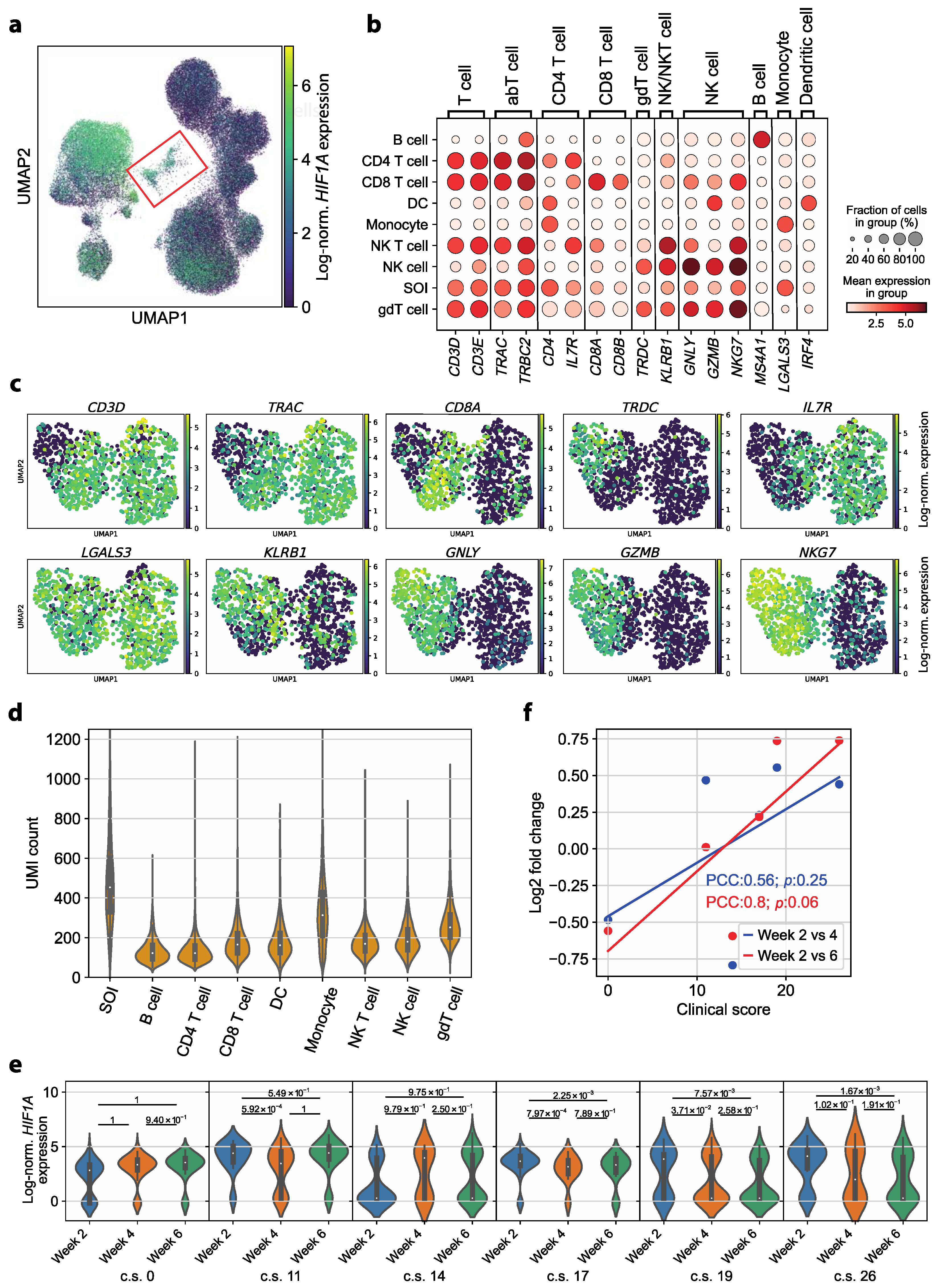
3.3. HIF1A Expression Is Significantly Upregulated in Monocytes of Symptomatic Patients Two Weeks after COVID-19 Disease Verification
3.4. HIF1A Upregulation during Convalescence Significantly Correlates with Previous COVID-19 Disease Severity Scores
3.5. Circulating Monocyte–T Cell and Monocyte–NK Cell Complexes Indicate a Persistent Dysregulation in Convalescent COVID-19 Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 18 December 2023).
- COVID-19 Epidemiological Update—24 November 2023. Available online: https://www.who.int/publications/m/item/covid-19-epidemiological-update---24-november-2023 (accessed on 18 December 2023).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, R.; Lu, Z.; Huang, Y. Does Comorbidity Increase the Risk of Patients with COVID-19: Evidence from Meta-Analysis. Aging (Albany N. Y.) 2020, 12, 6049–6057. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Y.; Lin, R.; Han, K. Clinical Features of COVID-19 in Elderly Patients: A Comparison with Young and Middle-Aged Patients. J. Infect. 2020, 80, e14–e18. [Google Scholar] [CrossRef] [PubMed]
- Espejo-Paeres, C.; Espliguero, R.A.; Uribarri, A.; Antón-Huguet, B.; Romero, R.; Fernández-Rozas, I.; Becerra-Muñoz, V.M.; Alfonso-Rodríguez, E.; Huang, J.; Ortega-Armas, M.E.; et al. Predictors of Poor Prognosis in Healthy, Young, Individuals with SARS-CoV-2 Infections. Clin. Microbiol. Infect. 2022, 28, 273–278. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Q.; Chen, J.; Xiang, R.; Song, H.; Shu, S.; Chen, L.; Liang, L.; Zhou, J.; You, L.; et al. Detection of Covid-19 in Children in Early January 2020 in Wuhan, China. N. Engl. J. Med. 2020, 382, 1370–1371. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Xu, Y.; Sun, C.; Wang, X.; Guo, Y.; Qiu, S.; Ma, K. A Systematic Review of Asymptomatic Infections with COVID-19. J. Microbiol. Immunol. Infect. 2021, 54, 12–16. [Google Scholar] [CrossRef]
- Liu, Q.; Qin, C.; Liu, M.; Liu, J. Effectiveness and Safety of SARS-CoV-2 Vaccine in Real-World Studies: A Systematic Review and Meta-Analysis. Infect. Dis. Poverty 2021, 10, 132. [Google Scholar] [CrossRef]
- Moghadas, S.M.; Vilches, T.N.; Zhang, K.; Wells, C.R.; Shoukat, A.; Singer, B.H.; Meyers, L.A.; Neuzil, K.M.; Langley, J.M.; Fitzpatrick, M.C.; et al. The Impact of Vaccination on Coronavirus Disease 2019 (COVID-19) Outbreaks in the United States. Clin. Infect. Dis. 2021, 73, 2257–2264. [Google Scholar] [CrossRef]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.-M.; et al. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Gómez-Rial, J.; Rivero-Calle, I.; Salas, A.; Martinón-Torres, F. Role of Monocytes/Macrophages in Covid-19 Pathogenesis: Implications for Therapy. Infect. Drug Resist. 2020, 13, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Wack, A. Monocyte and Dendritic Cell Defects in COVID-19. Nat. Cell Biol. 2021, 23, 445–447. [Google Scholar] [CrossRef]
- Guo, C.; Li, B.; Ma, H.; Wang, X.; Cai, P.; Yu, Q.; Zhu, L.; Jin, L.; Jiang, C.; Fang, J.; et al. Tocilizumab Treatment in Severe COVID-19 Patients Attenuates the Inflammatory Storm Incited by Monocyte Centric Immune Interactions Revealed by Single-Cell Analysis. bioRxiv 2020. bioRxiv:2020.04.08.029769. [Google Scholar]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A Single-Cell Atlas of the Peripheral Immune Response in Patients with Severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn Errors of Type I IFN Immunity in Patients with Life-Threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Ren, X.; Wen, W.; Fan, X.; Hou, W.; Su, B.; Cai, P.; Li, J.; Liu, Y.; Tang, F.; Zhang, F.; et al. COVID-19 Immune Features Revealed by a Large-Scale Single-Cell Transcriptome Atlas. Cell 2021, 184, 1895–1913.e19. [Google Scholar] [CrossRef]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association Between Hypoxemia and Mortality in Patients With COVID-19. Mayo Clin. Proc. 2020, 95, 1138–1147. [Google Scholar] [CrossRef]
- Ludvigsson, J.F. Case Report and Systematic Review Suggest That Children May Experience Similar Long-Term Effects to Adults after Clinical COVID-19. Acta Paediatr. 2021, 110, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-Acute COVID-19 Syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Therapeutic Trial Synopsis. Available online: https://www.who.int/publications-detail-redirect/covid-19-therapeutic-trial-synopsis (accessed on 18 December 2023).
- Mithal, A.; Jevalikar, G.; Sharma, R.; Singh, A.; Farooqui, K.J.; Mahendru, S.; Krishnamurthy, A.; Dewan, A.; Budhiraja, S. High Prevalence of Diabetes and Other Comorbidities in Hospitalized Patients with COVID-19 in Delhi, India, and Their Association with Outcomes. Diabetes Metab. Syndr. 2021, 15, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B. Python Implementation of ComBat. 2012. Available online: https://github.com/brentp/combat.py (accessed on 18 December 2023).
- Morrison, D.F. McGraw-Hill series in probability and statistics. In Multivariate Statistical Methods, 2nd ed.; McGraw-Hill: New York, NY, USA, 1976; ISBN 978-0-07-043186-7. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Traag, V.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- de Almeida, G.P.; Lichtner, P.; Eckstein, G.; Brinkschmidt, T.; Chu, C.-F.; Sun, S.; Reinhard, J.; Mädler, S.C.; Kloeppel, M.; Verbeek, M.; et al. Human Skin-Resident Host T Cells Can Persist Long Term after Allogeneic Stem Cell Transplantation and Maintain Recirculation Potential. Sci. Immunol. 2022, 7, eabe2634. [Google Scholar] [CrossRef]
- Kumari, S.; Nie, J.; Chen, H.-S.; Ma, H.; Stewart, R.; Li, X.; Lu, M.-Z.; Taylor, W.M.; Wei, H. Evaluation of Gene Association Methods for Coexpression Network Construction and Biological Knowledge Discovery. PLoS ONE 2012, 7, e50411. [Google Scholar] [CrossRef]
- Guan, W.; Liang, W.; Zhao, Y.; Liang, H.; Chen, Z.; Li, Y.; Liu, X.; Chen, R.; Tang, C.; Wang, T.; et al. Comorbidity and Its Impact on 1590 Patients with COVID-19 in China: A Nationwide Analysis. Eur. Respir. J. 2020, 55, 2000547. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single–Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Saichi, M.; Ladjemi, M.Z.; Korniotis, S.; Rousseau, C.; Ait Hamou, Z.; Massenet-Regad, L.; Amblard, E.; Noel, F.; Marie, Y.; Bouteiller, D.; et al. Single-Cell RNA Sequencing of Blood Antigen-Presenting Cells in Severe COVID-19 Reveals Multi-Process Defects in Antiviral Immunity. Nat. Cell Biol. 2021, 23, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Liu, W.; Li, X.; Zhao, P.; Shereen, M.A.; Zhu, C.; Huang, S.; Liu, S.; Yu, X.; Yue, M.; et al. HIF-1α Promotes SARS-CoV-2 Infection and Aggravates Inflammatory Responses to COVID-19. Sig. Transduct. Target. Ther. 2021, 6, 308. [Google Scholar] [CrossRef] [PubMed]
- Serebrovska, Z.O.; Chong, E.Y.; Serebrovska, T.V.; Tumanovska, L.V.; Xi, L. Hypoxia, HIF-1α, and COVID-19: From Pathogenic Factors to Potential Therapeutic Targets. Acta Pharmacol. Sin. 2020, 41, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Sieweke, M.; Geissmann, F. Blood Monocytes: Development, Heterogeneity, and Relationship with Dendritic Cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Martins, A.J.; Lau, W.W.; Rachmaninoff, N.; Chen, J.; Imberti, L.; Mostaghimi, D.; Fink, D.L.; Burbelo, P.D.; Dobbs, K.; et al. Time-Resolved Systems Immunology Reveals a Late Juncture Linked to Fatal COVID-19. Cell 2021, 184, 1836–1857.e22. [Google Scholar] [CrossRef] [PubMed]
- Gil-Manso, S.; Miguens Blanco, I.; López-Esteban, R.; Carbonell, D.; López-Fernández, L.A.; West, L.; Correa-Rocha, R.; Pion, M. Comprehensive Flow Cytometry Profiling of the Immune System in COVID-19 Convalescent Individuals. Front. Immunol. 2022, 12, 793142. [Google Scholar] [CrossRef]
- Burel, J.G.; Pomaznoy, M.; Lindestam Arlehamn, C.S.; Weiskopf, D.; da Silva Antunes, R.; Jung, Y.; Babor, M.; Schulten, V.; Seumois, G.; Greenbaum, J.A.; et al. Circulating T Cell-Monocyte Complexes Are Markers of Immune Perturbations. eLife 2019, 8, e46045. [Google Scholar] [CrossRef]
- Rowntree, D. Statistics Without Tears: A Primer For Non-Mathematicians; Penguin Books Ltd.: London, UK, 1981; ISBN 978-0-14-022326-2. [Google Scholar]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Bacher, R.; Kendziorski, C. Design and Computational Analysis of Single-Cell RNA-Sequencing Experiments. Genome Biol. 2016, 17, 63. [Google Scholar] [CrossRef]
- Zhao, P.; Zou, J.; Zhou, F.; Zhu, Y.; Song, Q.; Yu, D.; Li, X. Immune Features of COVID-19 Convalescent Individuals Revealed by a Single-Cell RNA Sequencing. Int. Immunopharmacol. 2022, 108, 108767. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, F.; Vazquez-Lombardi, R.; Yermanos, A.; Ehling, R.A.; Mason, D.M.; Wagner, B.; Kapetanovic, E.; Di Roberto, R.B.; Weber, C.R.; Savic, M.; et al. A Single-Cell Atlas of Lymphocyte Adaptive Immune Repertoires and Transcriptomes Reveals Age-Related Differences in Convalescent COVID-19 Patients. Front. Immunol. 2021, 12, 701085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Wang, X.-M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J.-W.; Fan, X.; Xia, P.; Fu, J.-L.; Wang, S.-Y.; et al. Single-Cell Landscape of Immunological Responses in Patients with COVID-19. Nat. Immunol. 2020, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The Fate and Lifespan of Human Monocyte Subsets in Steady State and Systemic Inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Lee, M.J.; Wei, B.; Parks, B.; Pi, R.; Martínez-Colón, G.J.; Ranganath, T.; Zhao, N.Q.; Taylor, S.; Becker, W.; et al. Multi-Omic Profiling Reveals Widespread Dysregulation of Innate Immunity and Hematopoiesis in COVID-19. J. Exp. Med. 2021, 218, e20210582. [Google Scholar] [CrossRef]
- Wang, X.; Wen, Y.; Xie, X.; Liu, Y.; Tan, X.; Cai, Q.; Zhang, Y.; Cheng, L.; Xu, G.; Zhang, S.; et al. Dysregulated Hematopoiesis in Bone Marrow Marks Severe COVID-19. Cell Discov. 2021, 7, 60. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.-L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-Converting Enzyme 2 (ACE2), SARS-CoV-2 and the Pathophysiology of Coronavirus Disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the Regulation ACE and ACE2 Expression in Hypoxic Human Pulmonary Artery Smooth Muscle Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Song, X.; Hu, W.; Yu, H.; Zhao, L.; Zhao, Y.; Zhao, Y. High Expression of Angiotensin-Converting Enzyme-2 (ACE2) on Tissue Macrophages That May Be Targeted by Virus SARS-CoV-2 in COVID-19 Patients. bioRxiv 2020. bioRxiv:2020.07.18.210120. [Google Scholar]
- Wang, Y.; Wang, Y.; Luo, W.; Huang, L.; Xiao, J.; Li, F.; Qin, S.; Song, X.; Wu, Y.; Zeng, Q.; et al. A Comprehensive Investigation of the mRNA and Protein Level of ACE2, the Putative Receptor of SARS-CoV-2, in Human Tissues and Blood Cells. Int. J. Med. Sci 2020, 17, 1522–1531. [Google Scholar] [CrossRef]
- Bosco, M.C.; Puppo, M.; Santangelo, C.; Anfosso, L.; Pfeffer, U.; Fardin, P.; Battaglia, F.; Varesio, L. Hypoxia Modifies the Transcriptome of Primary Human Monocytes: Modulation of Novel Immune-Related Genes and Identification Of CC-Chemokine Ligand 20 as a New Hypoxia-Inducible Gene. J. Immunol. 2006, 177, 1941–1955. [Google Scholar] [CrossRef]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte Adhesion during Hypoxia Is Mediated by HIF-1-Dependent Induction of Β2 Integrin Gene Expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
May, L.; Chu, C.-F.; Zielinski, C.E. Single-Cell RNA Sequencing Reveals HIF1A as a Severity-Sensitive Immunological Scar in Circulating Monocytes of Convalescent Comorbidity-Free COVID-19 Patients. Cells 2024, 13, 300. https://doi.org/10.3390/cells13040300
May L, Chu C-F, Zielinski CE. Single-Cell RNA Sequencing Reveals HIF1A as a Severity-Sensitive Immunological Scar in Circulating Monocytes of Convalescent Comorbidity-Free COVID-19 Patients. Cells. 2024; 13(4):300. https://doi.org/10.3390/cells13040300
Chicago/Turabian StyleMay, Lilly, Chang-Feng Chu, and Christina E. Zielinski. 2024. "Single-Cell RNA Sequencing Reveals HIF1A as a Severity-Sensitive Immunological Scar in Circulating Monocytes of Convalescent Comorbidity-Free COVID-19 Patients" Cells 13, no. 4: 300. https://doi.org/10.3390/cells13040300
APA StyleMay, L., Chu, C.-F., & Zielinski, C. E. (2024). Single-Cell RNA Sequencing Reveals HIF1A as a Severity-Sensitive Immunological Scar in Circulating Monocytes of Convalescent Comorbidity-Free COVID-19 Patients. Cells, 13(4), 300. https://doi.org/10.3390/cells13040300