1. Introduction
Coronaviruses are enveloped viruses with a single-stranded, positive-sense RNA genome of 26–32 kb in size [
1]. The genome encodes structural proteins including the spike (S), envelope (E), membrane (M), and nucleocapsid (N); accessory proteins 3, 6, 7a, 7b, 8, 9b, 10b, 13, and 14; and the large polyprotein 1ab (pp1ab). pp1ab is further processed into 16 individual non-structural proteins (NSPs) by papain-like proteases (NSP3) and 3C-like proteases (NSP5) for viral replication and transcription.
Coronaviruses infect primarily birds and mammals including humans. HCoVs such as HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 cause mild common cold [
2]. Since 2002, three new HCoVs causing severe disease have emerged—severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS), and SARS-CoV-2 [
3]. While outbreaks of SARS and MERS were relatively limited, SARS-CoV-2 caused a global pandemic for more than 3 years with more than 6.5 million deaths [
4]. Although the SARS-CoV-2 pandemic ended, SARS-CoV-2 evolution is still very much in progress as new variants continue to emerge and replace old ones [
5]. Given the extraordinarily efficient evolution of SARS-CoV-2, it is likely that new species of HCoV will emerge in the future.
Upon SARS-CoV-2 infection of nasal, bronchial, and lung epithelial cells, the innate immune response is initiated by the sensing of replication-intermediate double-stranded RNA by retinoic acid-inducible gene 1 (RIG-I) and anti-melanoma differentiation-associated gene 5 (MDA5) [
6,
7]. Due to proteolytic inactivation of RIG-I after the infection (see below), MDA5 activates the RIG-I-like receptor (RLR) signaling cascade, inducing the activation of two central transcription factors, interferon (IFN) regulatory factor 3 (IRF3) and NF-κB, leading to the transcriptional induction of IFN-β [
7]. IFN-β functions in both autocrine and paracrine manners to upregulate hundreds of IFN-stimulated genes that exert antiviral effects, either directly or indirectly through diverse mechanisms [
8,
9]. SARS-CoV-2 uses a variety of strategies to evade the IFN-mediated immune response and establish a productive infection by hijacking host proteins [
6]. One of the main strategies used for host immune evasion is the cleavage, and inactivation, of host proteins by SARS-CoV-2 NSP5. SARS-CoV-2 NSP5 cleaves RIG-I, eukaryotic translation initiation factor 4G (eIF4G), and NF-κB essential modulator (NEMO) to repress IFN-β production [
10,
11,
12]. In addition, SARS-CoV-2 NSP5 impairs inflammasome signaling by cleaving NOD-like receptor protein 1 (NLRP1) and NLRP12 [
13,
14].
TonEBP, also known as NFAT5 (nuclear factor of activated T cell 5), is a transcription factor of the Rel family with homology to NF-κB [
15]. TonEBP was initially discovered as a central regulator of cellular responses to hypertonic stress, a role it plays by binding to TonEs. Recent studies have revealed that TonEBP is a versatile stress protein associated with a viral infection. TonEBP is a proteolytic target of coxsackievirus B3 (CVB3) protease 2A [
16]. This cleavage promotes CVB3 replication and tissue damage due to the reduced expression of inducible nitric oxide synthase in cardiac myocytes. Thus, TonEBP is an antiviral protein against CVB3 infection in the heart, but it is disabled after CVB3 infection. On the other hand, TonEBP is a proviral protein for lymphocytic choriomeningitis virus replication [
17]. It engages with the IFN-β promoter in a Toll-like receptor 3-dependent manner, thereby hindering IRF3 binding and suppressing IFN-β promoter activity, ultimately dampening innate immune responses. Here, we find that NSP5 from several HCoVs cleaves TonEBP. One of the cleavage products, TonEBP NT directly binds to the IFN-β promoter and suppresses
IFN-β gene expression.
2. Materials and Methods
2.1. Cell Culture and Transfection
A549 (CCL-185), HEK293T (CRL-1573), U2OS (HTB-96), and Vero-E6 (CRL-1586) cells were obtained from the American Type Culture Collection (Manassas, VA, USA). HEK293T, U2OS, and Vero-E6 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), whereas A549 cells were cultured in Roswell Park Memorial Institute-1640 medium with 25 mM HEPES and 10% FBS, at 37 °C, in an incubator with 5% CO2. For transient transfection, HEK293T cells were transfected at 70% confluence using jetPRIME® (Polyplus-transfection, Illkirch-Graffenstaden, France) according to the manufacturer’s instructions.
2.2. Plasmids
The TonE-driven Photinus luciferase plasmid has been described previously [
15]. The Renilla luciferase plasmid (pRL-TK) was purchased from Promega (Madison, WI, USA). Plasmids expressing individual SARS-CoV-2 NSPs were cloned by gateway cloning, using the corresponding donor vector (#141255–#141269, Addgene, Watertown, MA, USA) and a CMV-promoter-driven expression vector. Transient expression plasmids of NSP5s from the 229E, NL63, OC43, HKU9, and SARS-CoV-2 strains have been reported previously [
18]. To compare SARS-CoV-2 NSP5 wild type and C145A mutant, EF-1 alpha-promoter-driven expression vector (#141370, #141371) were purchased from Addgene.
The plasmid encoding His-SARS-CoV-2 NSP5 was obtained from Addgene (m4133256). For substitution mutations in TonEBP, stie-directed mutagenesis was carried out using KOD Plus (KOD-201, TOYOBO, Osaka, Japan), according to the manufacturer’s instructions. The plasmid encoding the N-terminal fragment of TonEBP generated by SARS-CoV-2 NSP5 cleavage was cloned into the pCMV-tag 2B vector (#211172, Agilent Technologies, Santa Clara, CA, USA) by amplification and standard restriction cloning.
The IFN-β promoter luciferase reporter (#102597), as well as transient expression vectors for MDA5 (#52876), MAVS (#52135), TBK1 (#131792), IRF3 (#127663), and p65 (#111192), were obtained from Addgene. For generation of the expression vector for IRF3-5D, site-directed mutagenesis was performed using KOD Plus, according to the manufacturer’s instructions.
2.3. Viral Infection
The human coronavirus HCoV-OC43 (betacoronavirus 1, KBPV-VR-8) was obtained from Korea Bank for Pathogenic Viruses (Seoul, Republic of Korea) and stored at −80 °C. Viral infection titers were measured using a focus-forming assay on Vero-E6 cells. Vero-E6 cells were grown to confluence in 96-well plates. Cells at 90% confluence were infected with 10-fold serial dilutions of a HCoV-OC43 stock solution for 3 h at 33 °C in an incubator maintained at 5% CO2 without serum and with gentle rocking. After infection, the cells were cultured for 24 h in regular medium supplemented with 2% FBS. The cells were then fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton™ X-100. After blocking with 3% bovine serum albumin, immunofluorescence was performed using a mouse monoclonal antibody against HCoV-OC43 N protein (MAB9012, Merck Millipore, Burlington, MA, USA). Antibody labeling was detected using a secondary antibody conjugated with Alexa Fluor™ 633 (Invitrogen, Carlsbad, CA, USA). Foci were visualized and counted using a fluorescence microscope (IN Cell Analyzer 2500, Cytiva, Marlborough, MA, USA).
A549, HEK293T, and U2OS cells were incubated with HCoV-OC43 for 3 h at different multiplicities of infection without serum. After removing the infectious solution, the cells were washed with serum-free medium and maintained in regular medium containing 2% FBS. The infected cells were subjected to Western blot, luciferase assays, or immunofluorescence.
2.4. Immunoblot Analysis and Immunoprecipitation
For immunoblot analysis, the cells were washed three times with cold PBS and then lysed with an appropriate volume of 0.1% sodium dodecyl sulfate (SDS) lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 1% Triton™ X-100). After rotation at 4 °C for 10 min, the lysates were centrifuged at 13,000 rpm and 4 °C for 10 min, following which the supernatant was collected. SDS dye (4×; 0.2 M Tris-HCl pH 6.8, 8% SDS, 40% glycerol, and 0.04% bromophenol blue) was then added to the supernatant and incubated at 95 °C for 10 min.
For immunoprecipitation, the cells were washed three times with cold PBS and then lysed with an appropriate volume of lysis buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, and 1% NP-40). After rotation at 4 °C for 10 min, the lysates were centrifuged at 13,000 rpm and 4 °C for 10 min and the supernatant was incubated at 4 °C with anti-Flag M2 agarose beads (A2220, Merck Millipore) overnight. SDS dye (4×; 0.2 M Tris-HCl pH 6.8, 8% SDS, 40% glycerol, and 0.04% bromophenol blue) was then added to the lysates and the immunoprecipitated samples were incubated at 95 °C for 5 min.
Samples were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride or nitrocellulose membranes. The membranes were blocked with Tris-buffered saline + 0.1% Tween 20 supplemented with 7% skim milk for 1 h. After blocking, the membranes were incubated overnight with the following primary antibodies: rabbit anti-TonEBP (1:3000 in blocking solution) [
15]; monoclonal mouse anti-HCoV-OC43 N protein (1:1000 in blocking solution; MAB9012) from Merck Millipore; monoclonal mouse anti-β-actin (1:1000 in blocking solution; SC-47778) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); polyclonal rabbit anti-GAPDH (1:10,000 in blocking solution; 10494-1-AP), monoclonal mouse anti-GFP (1:10,000 in blocking solution; 66002-1-Ig), and monoclonal mouse anti-His (1:10,000 in blocking solution; 66005-1-Ig) from ProteinTech (Rosemont, IL, USA); monoclonal mouse anti-Flag (1:1000 in blocking solution; F1804) from Sigma-Aldrich (St. Louis, MO, USA); and monoclonal rabbit anti-TBK1/NAK (1:1000 in blocking solution; #3504), anti-phospho-TBK1/NAK (1:1000 in blocking solution; #5483), anti-IRF3 (1:1000 in blocking solution; #4302), and anti-phospho-IRF3 (1:1000 in blocking solution; #29047) from Cell Signaling Technology (Danvers, MA, USA). After several washes with Tris-buffered saline + 0.1% Tween 20, each blot was further incubated with the appropriate secondary antibody, either goat anti-mouse (1:10,000 in blocking solution; AP124P) or anti-rabbit (1:10,000 in blocking solution; AP307P) IgG conjugated to horseradish peroxidase (both from Millipore). The polyvinylidene difluoride blots were detected via chemiluminescence using Clarity™ Western ECL substrate solution (Bio-Rad, Hercules, CA, USA), whereas nitrocellulose blots were incubated with fluorescence-conjugated secondary antibodies: IRDye
® 800CW goat anti-rabbit (1:10,000 in blocking solution; 926-32211) or 680RD goat anti-mouse (1:10,000 in blocking solution; 926-68070) (LI-COR, Lincoln, NE, USA). Fluorescent blot detection was performed using an Odyssey
® CLx Imaging System (LI-COR).
2.5. Luciferase Assay
HEK293T cells were co-transfected with the indicated luciferase constructs of TonE-driven Photinus [
15], or
IFN-β promoter luciferase reporter (#102597, Addgene) and the Renilla luciferase plasmid (pRL-TK), which was used as an internal control for transfection efficiency. For the
IFN-β promoter luciferase assay, control plasmid or plasmids expressing MDA5, MAVS, TBK1, IRF3-5D, or p65 were used. Luciferase assays were performed using the Dual-Luciferase
® Reporter Assay System (Promega). For each condition, the luciferase activity was measured using samples taken from triplicate wells using a 96-well automated luminometer (Bio-Rad). Results were calculated as the ratio of firefly luciferase activity to Renilla luciferase activity.
2.6. Sequence Alignment
NSP5 protein sequences of coronaviruses were derived from available ORF1ab sequences: 229E (NP_073549.1), NL63 (YP_003766.2), OC43 (YP_009555238.1), HKU9 (YP_001039970.1), and SARS-CoV-2 (YP_009724389.1). These NSP5 protein sequences were aligned and analyzed using SnapGene version 7.2.1 (GSL Biotech LLC, San Diego, CA, USA).
2.7. Immunofluorescence Staining
HEK293T cells were seeded onto a 4-well chamber (#177429, Thermo Scientific, Waltham, MA, USA) and transfected with the indicated plasmids. The transfected cells were incubated at 37 °C for 48 h and then fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton™ X-100, and blocked with 3% bovine serum albumin for 1 h. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). The cells were observed under a Zeiss LSM 780 confocal microscope (Carl Zeiss, Jena, Germany).
2.8. U2OS Stable Cell Line Construction
HEK293T cells were seeded into 6-well plates at a density of 2 × 105 cells/mL and then transfected with lentiviral constructs (YFP and YFP-TonEBP NT) and packaging plasmids (VSVg, p8.2). After transfection for 16 h, the cells were washed with PBS and fresh medium was added. After 48 h of transfection, the lentiviral supernatants were collected and used to treat U2OS cells. After 48 h of lentiviral infection, U2OS cells were subjected to treatment with 2 μg/mL puromycin to select single-cell colonies for further experiments. Lentiviral constructs were cloned into the pLIX403-EYFP-ccdB-Puro plasmid (#158549; Addgene).
2.9. RNA Extraction and Quantitative Real-Time PCR
Total cellular RNA was extracted by phenol–chloroform extraction using RiboEX™ (GeneAll, Seoul, Republic of Korea), according to the manufacturer’s instructions. A total of 1 µg of RNA was converted into cDNA by reverse transcription using the ReverTra Ace-α® cDNA Synthesis Kit (TOYOBO). mRNA expression levels were detected by quantitative real-time PCR using SYBR® Green PCR Master Mix (TOYOBO). Actin mRNA was used as an endogenous control. All experiments were performed in triplicate. The primers used are given below:
Human IFN-β forward, AAACTCATGAGCAGTCTGCA;
Human IFN-β reverse, AGGAGATCTTCAGTTTCGGAGG;
Human GAPDH forward, CTGAACGGGAAGCTCACTGGCATG;
Human GAPDH reverse, AGGTCCACCACCCTGTTGCTGTAGC;
HCoV-OC43 Nucleocapsid forward, CGATGAGGCTATTCCGACTAGGT;
HCoV-OC43 Nucleocapsid reverse, CCTTCCTGAGCCTTCAATATAGTAACC.
2.10. Extracellular OC43 Titer Assay
To define the standard curve for the extracellular HCoV-OC43 titer, we extracted viral RNA from 150 µL of 10-fold serial dilutions of the HCoV-OC43 stock using the PURY Viral RNA Mini Kit (GenDEPOT, Katy, TX, USA). The cDNAs of viral RNA were synthesized by reverse transcription using the ReverTra Ace-α® cDNA Synthesis Kit, with random primers. The synthesized cDNAs was analyzed using quantitative real-time PCR, and the Cq values for each titer were used to define the standard curve. For the viral titer assay, 150 µL of extracellular medium containing HCoV-OC43 was collected at 68 h post-infection. Viral RNA was isolated, converted into cDNA, and analyzed using quantitative real-time PCR. Cq values were used to calculate the extracellular viral titers using a standard curve.
2.11. Focus-Forming Assay
Cells were seeded at a density of 2 × 105 cells/mL and then treated with doxycycline the following day. After 24 h of induction of protein expression, the cells were infected with the OC43 virus at a multiplicity of infection of 0.001. After 2 h of infection, the cells were washed more than three times with PBS, following which 2% FBS containing 1% low-melting agarose was added to cover the surface. A 10% paraformaldehyde solution was added above the agarose layer and cells were fixed for more than 1 h at 24 h post-infection. After fixation, the agarose layer was removed and the cells were permeabilized with 0.1% Triton™ X-100 for 10 min. Cells were blocked with 3% bovine serum albumin for 2 h and then incubated overnight with a primary antibody against OC43 N protein (1:1000 in blocking solution; MAB9012). After several washes with PBS, the cells were incubated with a fluorescence-conjugated secondary antibody (1:1000 in blocking solution; anti-mouse Alexa Fluor™ 594; A-11032; Thermo Fisher Scientific, Waltham, MA, USA) for 2 h. Nuclear staining was performed using Hoechst solution for 10 min. The number of OC43-infected cells per foci was analyzed using an IX83 inverted microscope (Olympus, Tokyo, Japan).
2.12. Enzyme-Linked Immunosorbent Assay
The IFN-β concentrations in HEK293T cell culture supernatants were determined using a Human IFN-β Assay Kit (DIFNB0, R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions. The absorbance was recorded at a wavelength of 450 nm using a Tecan infinite™ M200 plate reader (Tecan, Männedorf, Switzerland), with wavelength correction by subtracting the absorbance at 540 nm.
2.13. Chromatin Immunoprecipitation (ChIP) Analysis
For ChIP analysis, cells were crosslinked with 1% formaldehyde (F8775; Sigma-Aldrich) for 10 min and quenched with 0.125 M glycine for 5 min, at room temperature. ChIP was performed using a ChIP Assay Kit (17-295; Sigma-Aldrich), according to the manufacturer’s instructions. Antibodies against TonEBP [
15], and normal rabbit IgG (2729; Cell Signaling Technology) were used. Input and immunoprecipitated DNA were purified using a QIAquick
® PCR Purification Kit (Qiagen, Hilden, Germany). The purified input and immunoprecipitated DNA were detected by real-time PCR, using specific primers. The immunoprecipitated DNA from each sample was calculated as a percentage of the respective chromatin input and normalized to the control experiment for each condition. The following primers were used:
IFN-β promoter forward, AGGACCATCTCATATAAATAGGCCATACCC;
IFN-β promoter reverse, ACTGAAAATTGCTGCTTCTTTGTAGGAATC.
2.14. Protein Expression and Purification
Recombinant TonEBP sNT or p65 was expressed in the N-terminal His-fused form in E. coli BL21 cells (DE3). BL21 transformed with the expression plasmid of TonEBP sNT or p65 were grown in 2.0 L Luria Broth supplemented with the appropriate antibiotics, at 37 °C, with shaking, until the optical density at the wavelength of 600 nm reached a value of ~0.6. The BL21 culture was then cooled to 18 °C, and protein expression was induced by addition of 1.0 mM isopropyl β-D-1-thiogalactopyranoside. The cells were harvested after induction for 20 h by centrifugation at 4000× g, 4 °C, for 20 min.
The cells were resuspended in pre-cooled buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, and 1 mM dithiothreitol (DTT). The resuspended cells were then lysed by sonication for 9 cycles (20 s on and 30 s off) at an amplitude of 35, on ice, following which the cell debris were removed by centrifugation at 17,000× g and 4 °C for 1 h. The resulting supernatant was supplemented with 1 mL pre-equilibrated HisPur™ Ni-NTA resin (Thermo Scientific) and 20 mM imidazole and incubated overnight at 4 °C. Following that, the samples were loaded onto a gravity column and washed three times with 10 mL of wash buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 20 mM imidazole, and 1 mM DTT. The protein was then eluted in 1 mL of elution buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 200 mM imidazole, and 1 mM DTT. The eluted proteins were loaded onto a Superdex™ 75 Increase 10/300 GL (29-1487-21; Cytiva) gel filtration column equilibrated with PBS pH 7.4. The protein samples were finally aliquoted and stored at –80 °C.
2.15. Electrophoretic Mobility Shift Assay (EMSA)
The indicated amounts of purified His-TonEBP sNT or -p65 and 100 nM of indicated Cy5.5-conjugated probe were incubated at room temperature in the dark for 25 min, in a total volume of 20 μL in binding buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 40 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1 mM DTT, and 50 μg/mL bovine serum albumin). In competition assays, p65 was incubated with probe for 20 min prior to incubation with TonEBP sNT for 25 min. After incubation, the resulting mixtures were subjected to electrophoresis on a 0.8% agarose gel in 0.5× Tris-borate-EDTA (45 mM Tris, 45 mM borate, 1 mM EDTA pH 8.3). The gel was exposed and imaged using the Odyssey® CLx Imaging System (Li-COR). The following probes were used:
Probe IRF3/p65, Cy5.5-ATAGGAAAACTGAAAGGGAGAAGTGAAAGTGGGAAATTCC;
Probe IRF3, Cy5.5-GAAAACTGAAAGGGAGAAGTGAAAGTG;
Probe p65, Cy5.5-AATGTAAGTGGGAAATTCCTCTGAATA.
2.16. Surface Plasmon Resonance Assay
The affinities between TonEBP sNT or p65 and IFN-β promoter were measured at room temperature using a Biacore™ T200 system (Cytiva) with a streptavidin-coated sensor chip, Series S Sensor Chip SA (BR100531, Cytiva). The biotin-tagged IFN-β promoter probe was immobilized on the chip with a concentration of 10 nM and at a flow rate of 10 μL/min. A blank channel was left untreated. The same procedure was used to immobilize biotin as a negative control. For all measurements, the same running buffer, consisting of 10 mM HEPES, 150 mM NaCl, 3 mM EDTA, and 0.05% Surfactant P20, was used. Serially diluted recombinant TonEBP sNT or p65 was then allowed to flow through the chip surface, at a flow rate of 50 μL/min. Data were analyzed using the T200 BIA evaluation software (GE Healthcare, Arlington Heights, IL, USA), with curve fitting to the data obtained using the 1:1 binding model. The following probes were used:
Probe IRF3/p65, Biotin-ATAGGAAAACTGAAAGGGAGAAGTGAAAGTGGGAAATTCC;
Probe IRF3, Biotin-GAAAACTGAAAGGGAGAAGTGAAAGTG;
Probe p65, Biotin-AATGTAAGTGGGAAATTCCTCTGAATA.
2.17. Statistics
Statistical analyses were performed using Prism version 9 (GraphPad Software, San Diego, CA, USA).
4. Discussion
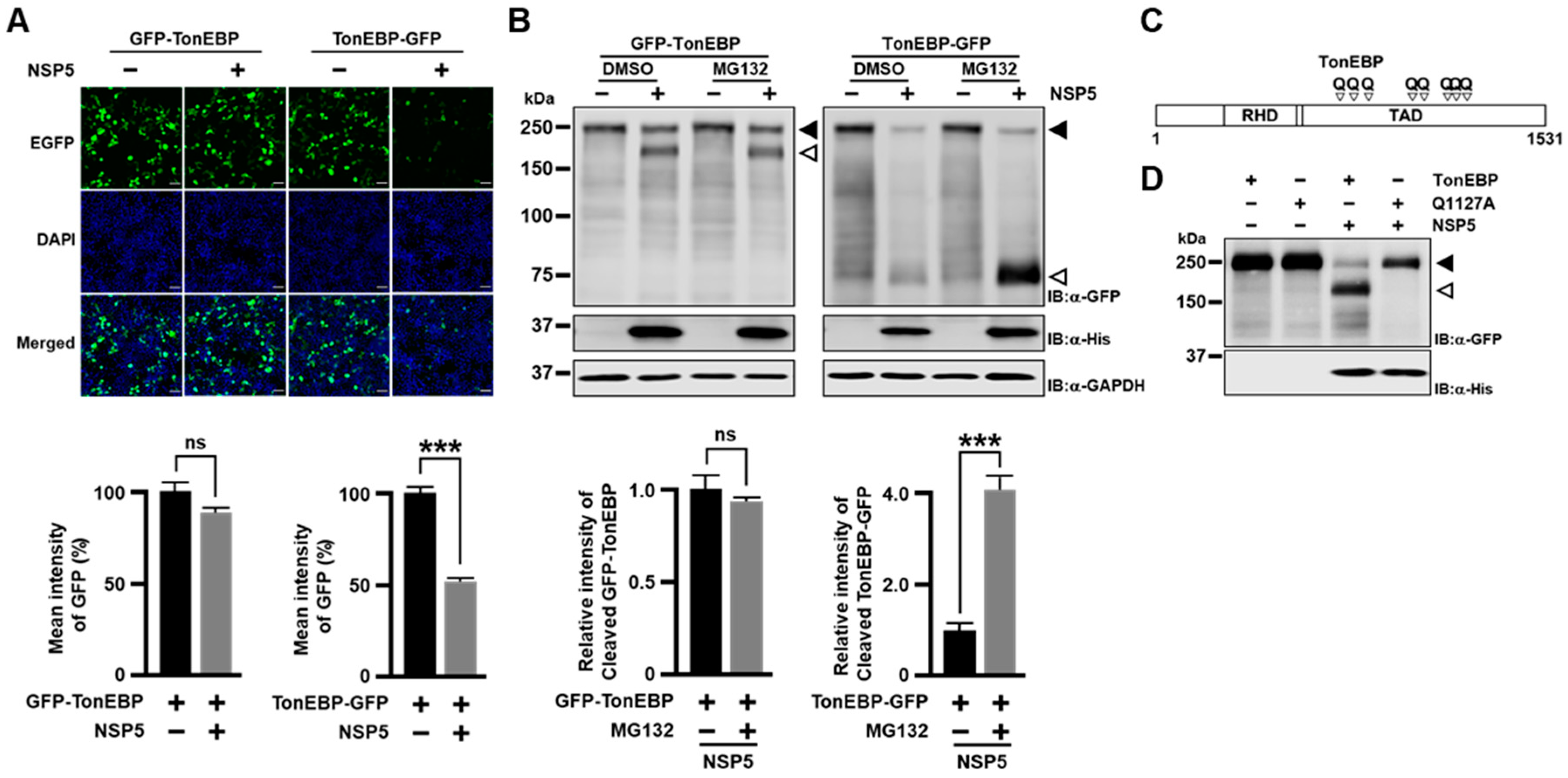
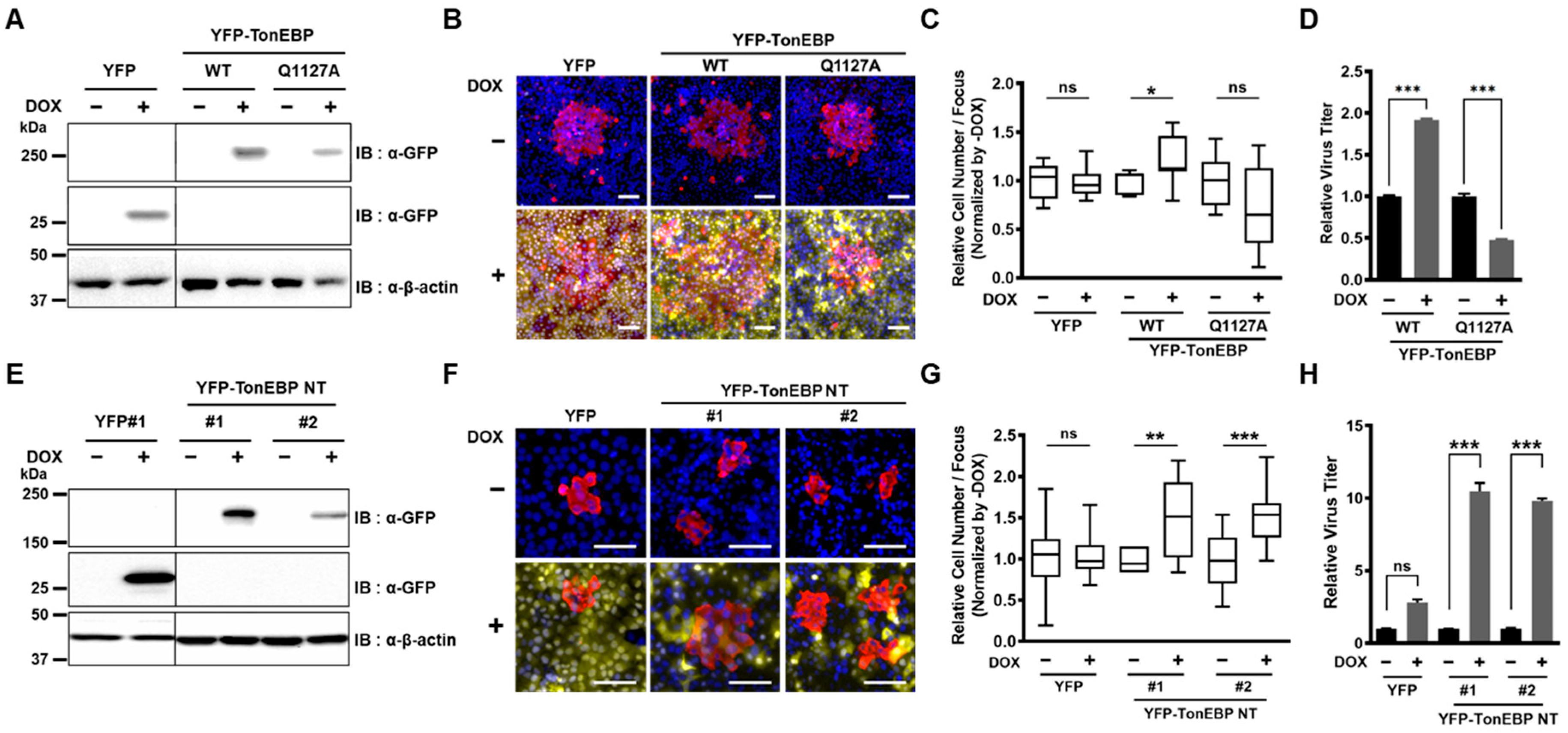
The data presented here demonstrate that NSP5s from HCoVs cleave TonEBP at amino acid position 1127 producing TonEBP NT. While TonEBP NT is incapable of enhancing the TonE-dependent transcription, it gains a new function to suppress the viral induction of IFN-β and, as a result, promote expansion of HCoV-OC43. A cleavage-resistant Q1127A mutant of TonEBP suppresses viral expansion in association with elevated IFN-β response (see more below). TonEBP adds to the repertoire of NSP5 targets to suppress the IFN-β induction by viral infection—RIG-I, eIF4G and NEMO [
10,
11,
12].
How TonEBP NT gains the ability to suppress the IFN-β response is of great interest. Evidence for this new function is 2 fold. First is differential effects of TonEBP vs. a cleavage-resistant Q1127A mutants in viral expansion (
Figure 3A–D) and IFN-β response (
Figure 4G,H). The other is the dramatic effects of TonEBP NT in promoting viral expansion (
Figure 3E–H) and suppressing the IFN-β response (
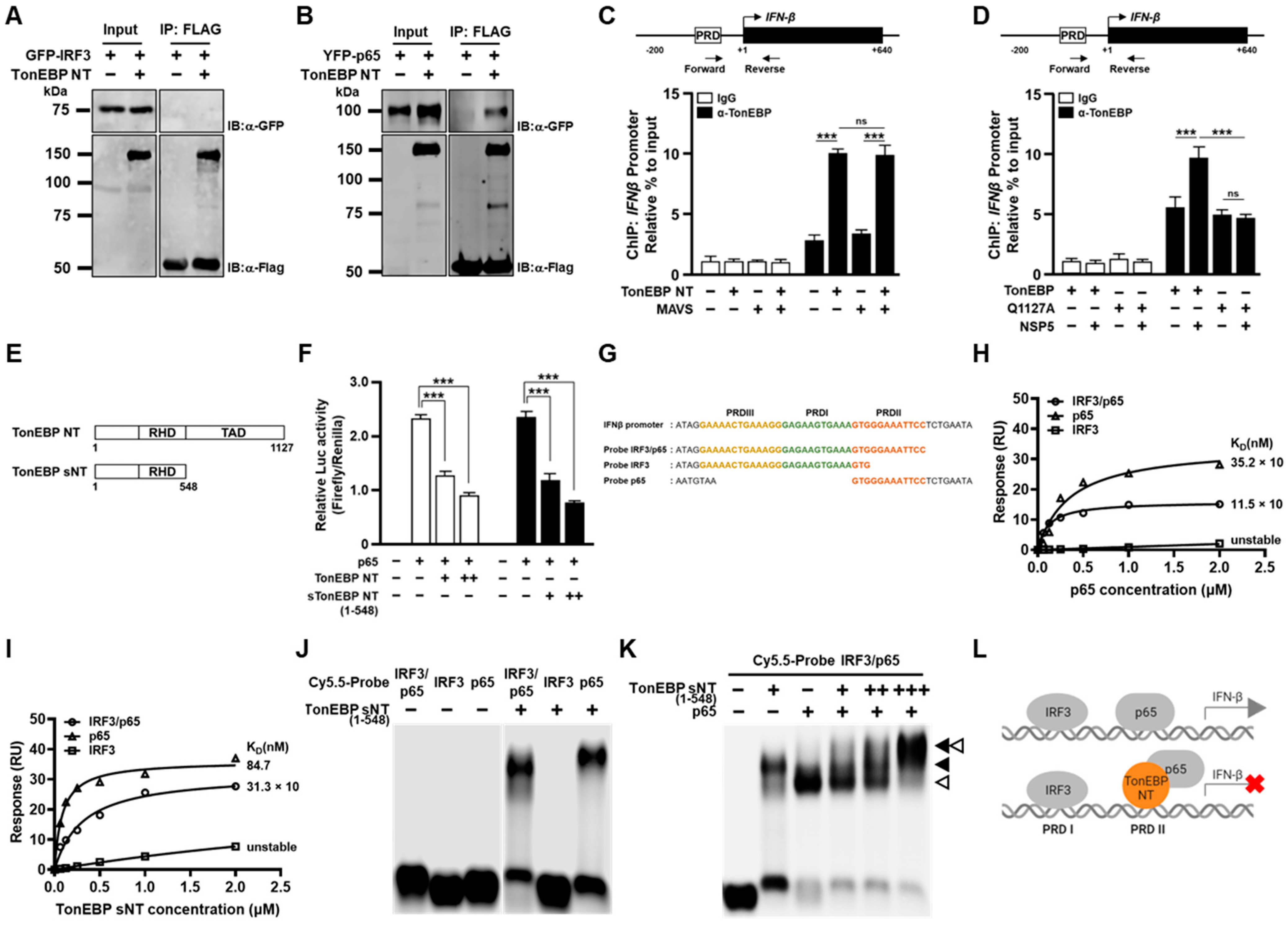
Figure 4A–E). Biochemical analyses of TonEBP NT suggest that the cleavage confers the ability to suppress the transcriptional activity of NF-κB (p65) perhaps by competing away DNA binding (
Figure 5K,L). This model is supported by elevated binding of TonEBP or TonEBP NT, but not by the Q1127A TonEBP, to the
IFN-β promoter, which was measured using antibodies that detect both TonEBP NT and full-length TonEBP (
Figure 5C,D).
TonEBP is also cleaved by CVB3 protease 2A at position 579 producing a protein similar to TonEBP sNT (see
Figure 5E) [
16]. This cleavage removes the entire TAD and inactivates the TonE-dependent transcriptional stimulation by TonEBP. As a result, inducible nitric oxide synthase expression is suppressed, leading to enhanced CVB3 replication and cardiac tissue damage because nitric oxide inhibits CVB3 replication by inactivating viral protease 2A and 3C [
21]. Since the cardiac tissues are capable of IFN-β induction in response to CVB3 infection [
22], we suspect that the N-terminal TonEBP fragment would also suppress the IFN-β induction like TonEBP sNT would (see
Figure 5F) in the heart after CVB3 infection. It is likely that the cleavage of TonEBP promotes the viral expansion using multiple cellular pathways in the heart.
In macrophages and dendritic cells, infection by vesicular stomatitis virus or murine cytomegalovirus results in elevated TonEBP expression [
17]. These viruses do not have genes encoding proteases. The elevated TonEBP, in turn, suppresses the IFN-β response by interfering with IRF3 binding. We also observed this effect when TonEBP was overexpressed (
Figure 5G,H). Thus, TonEBP by itself (i.e., when it is not cleaved) is a proviral factor; myeloid deficiency of TonEBP is associated with elevated IFN-β response and reduced viral load in response to infection by lymphocytic choriomeningitis virus, which also does not encode a protease [
17]. When infected by HCoV-OC43, TonEBP disappears in a time- and dose-dependent manner (
Figure 1B,C). The disappearance of TonEBP should lead to elevated IFN-β response and suppression of viral expansion. But the cleavage of TonEBP produces TonEBP NT, which is another powerful anti-IFN-β factor with a new mechanism targeting NF-κB. The gain of function of TonEBP after the proteolytic cleavage compensates for the loss of full-length TonEBP to support viral expansion. Since this mechanism appears common to all the HCoVs tested, it is likely to be relevant to new variants and species of HCoVs that emerge in the future.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}