
Investigation into Cardiac Myhc-α 334–352-Specific TCR Transgenic Mice Reveals a Role for Cytotoxic CD4 T Cells in the Development of Cardiac Autoimmunity

 , , , , and
, , , , and 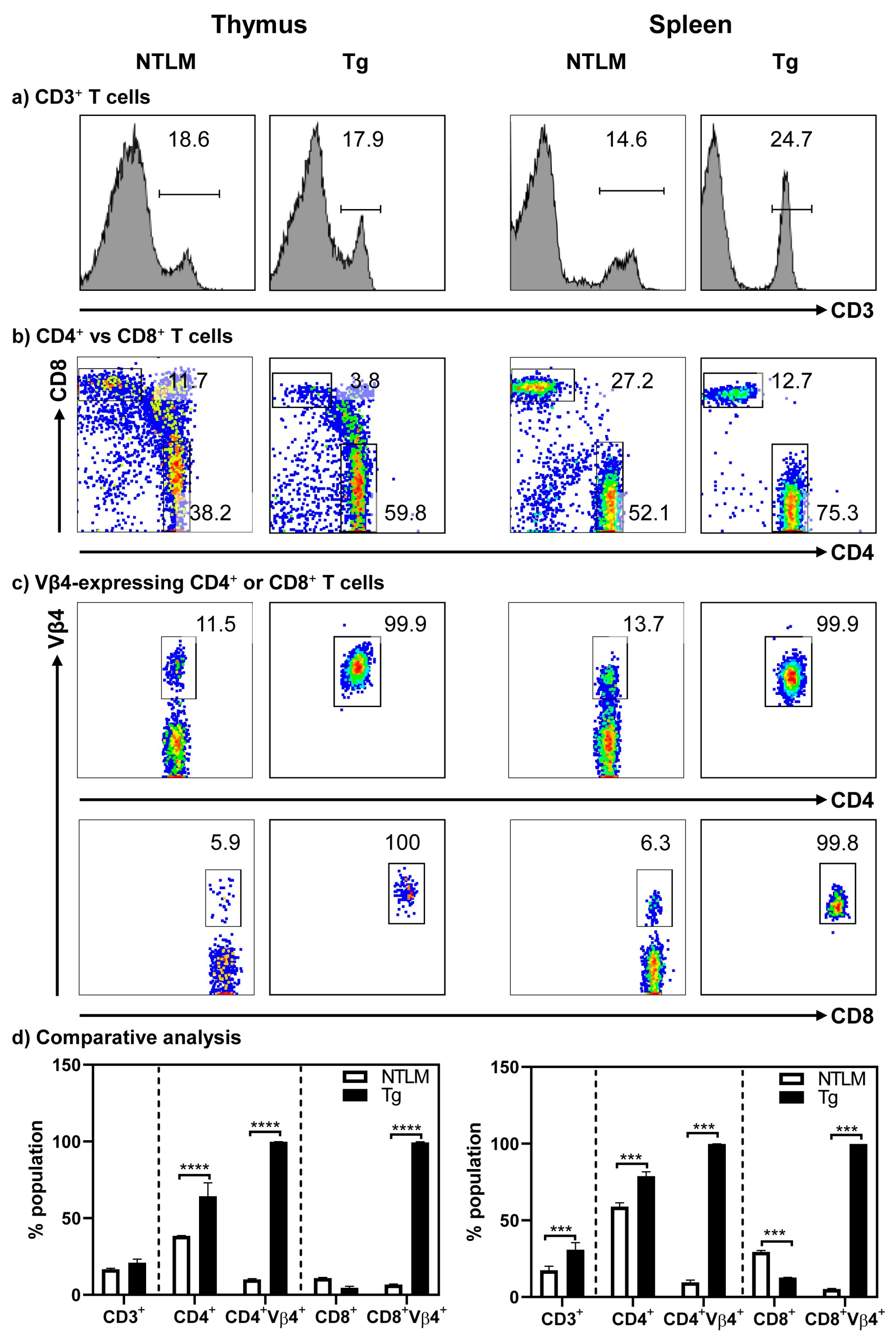{kind=link}
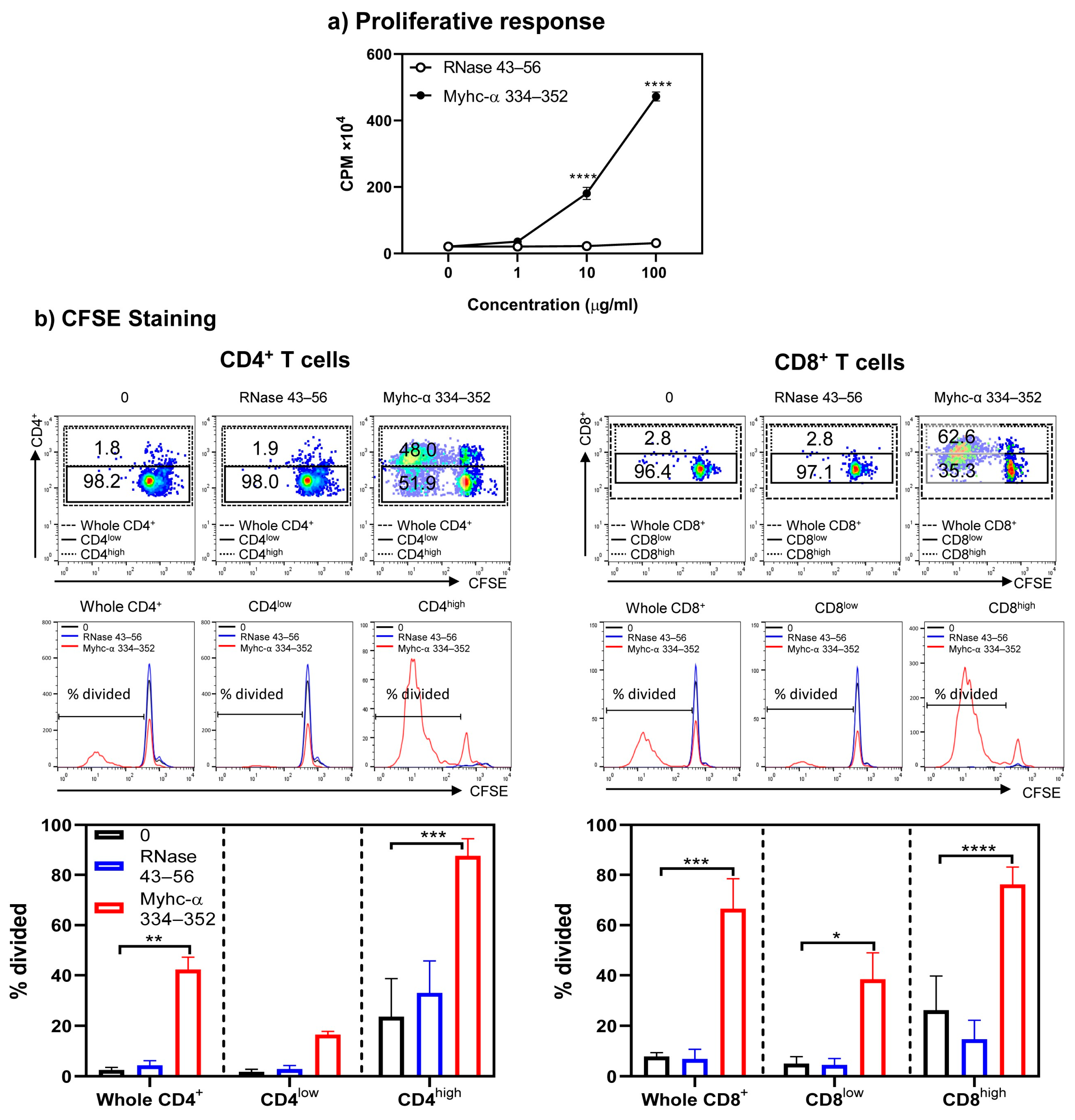{kind=link}
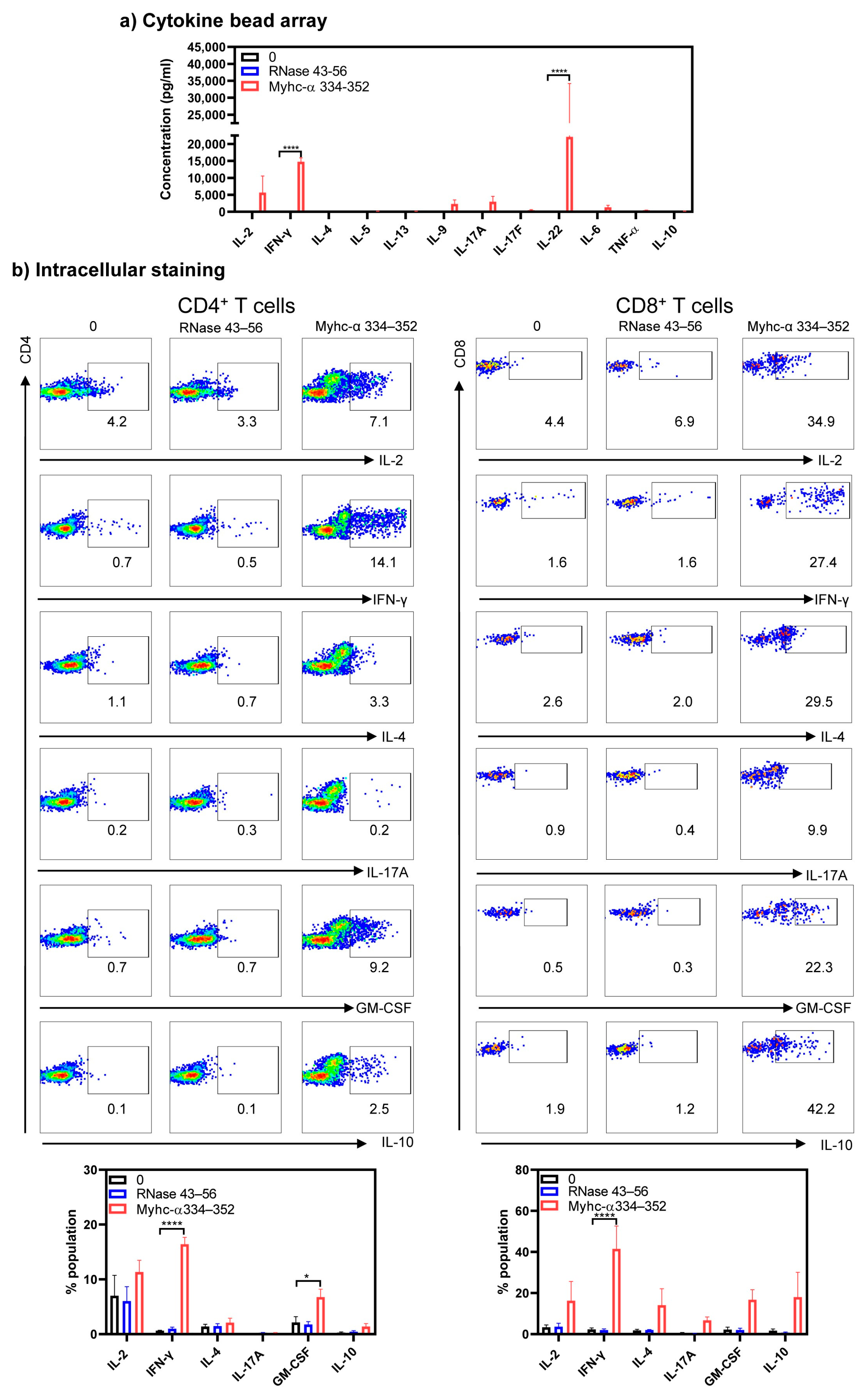{kind=link}
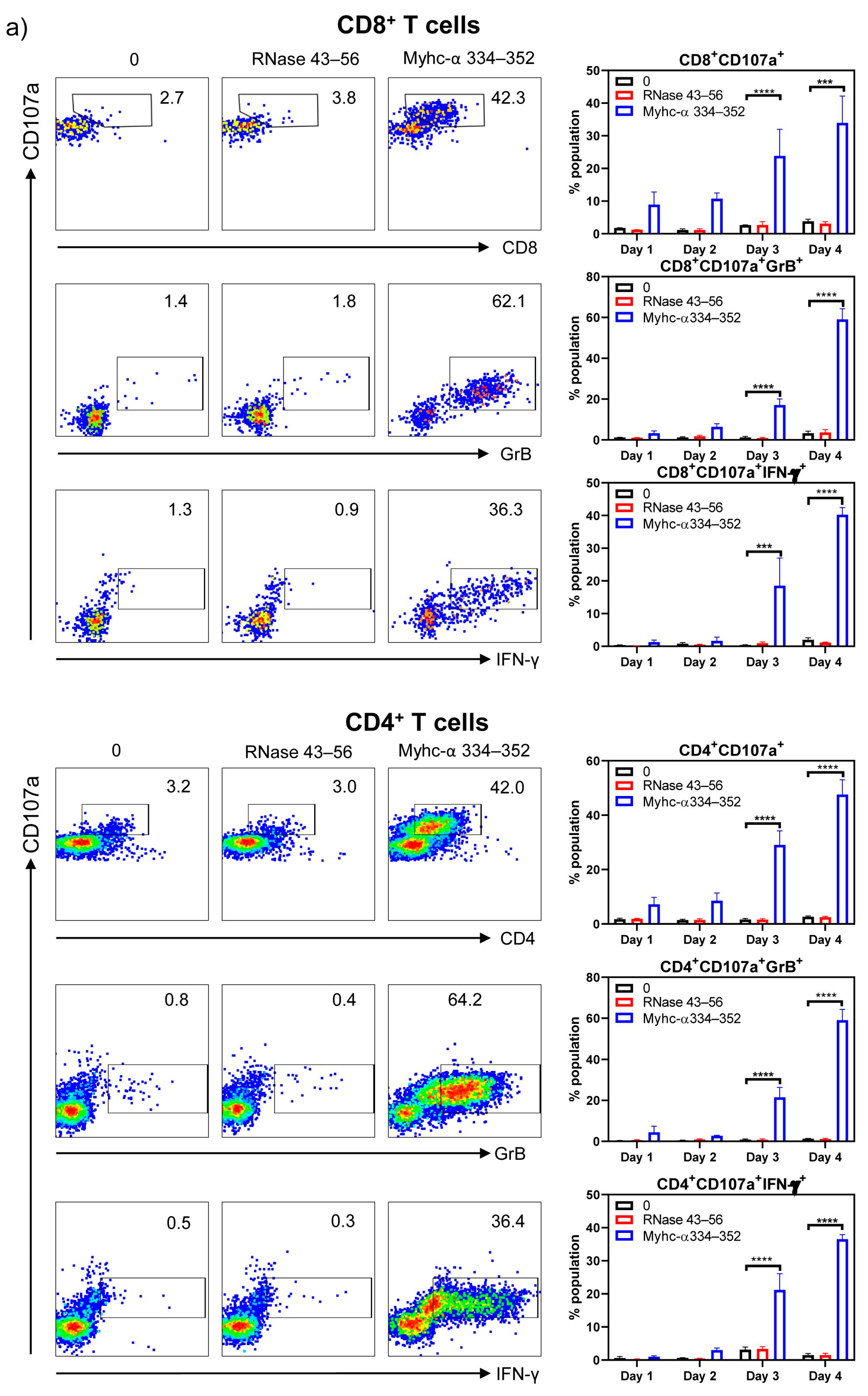{kind=link}
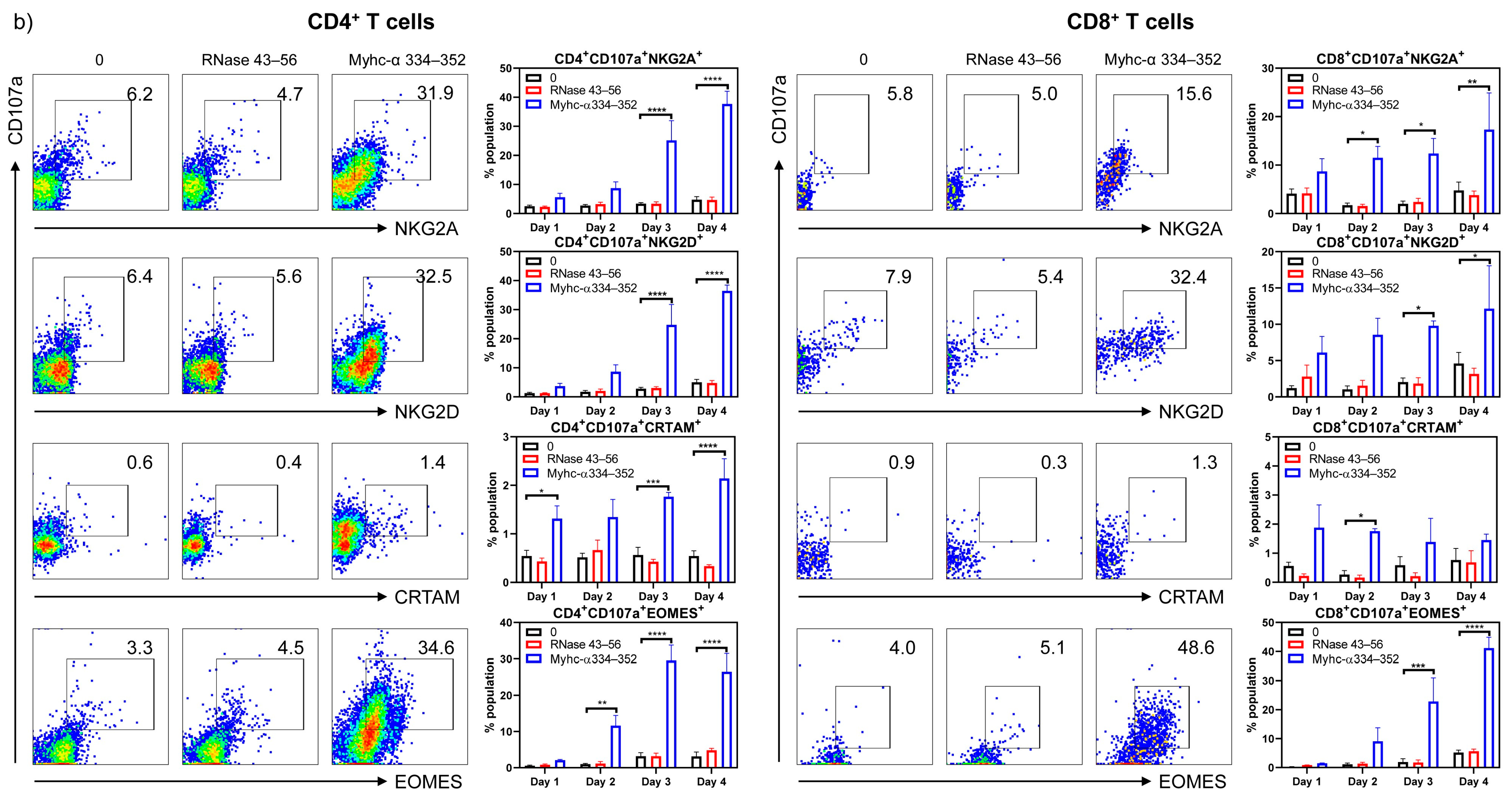{kind=link}
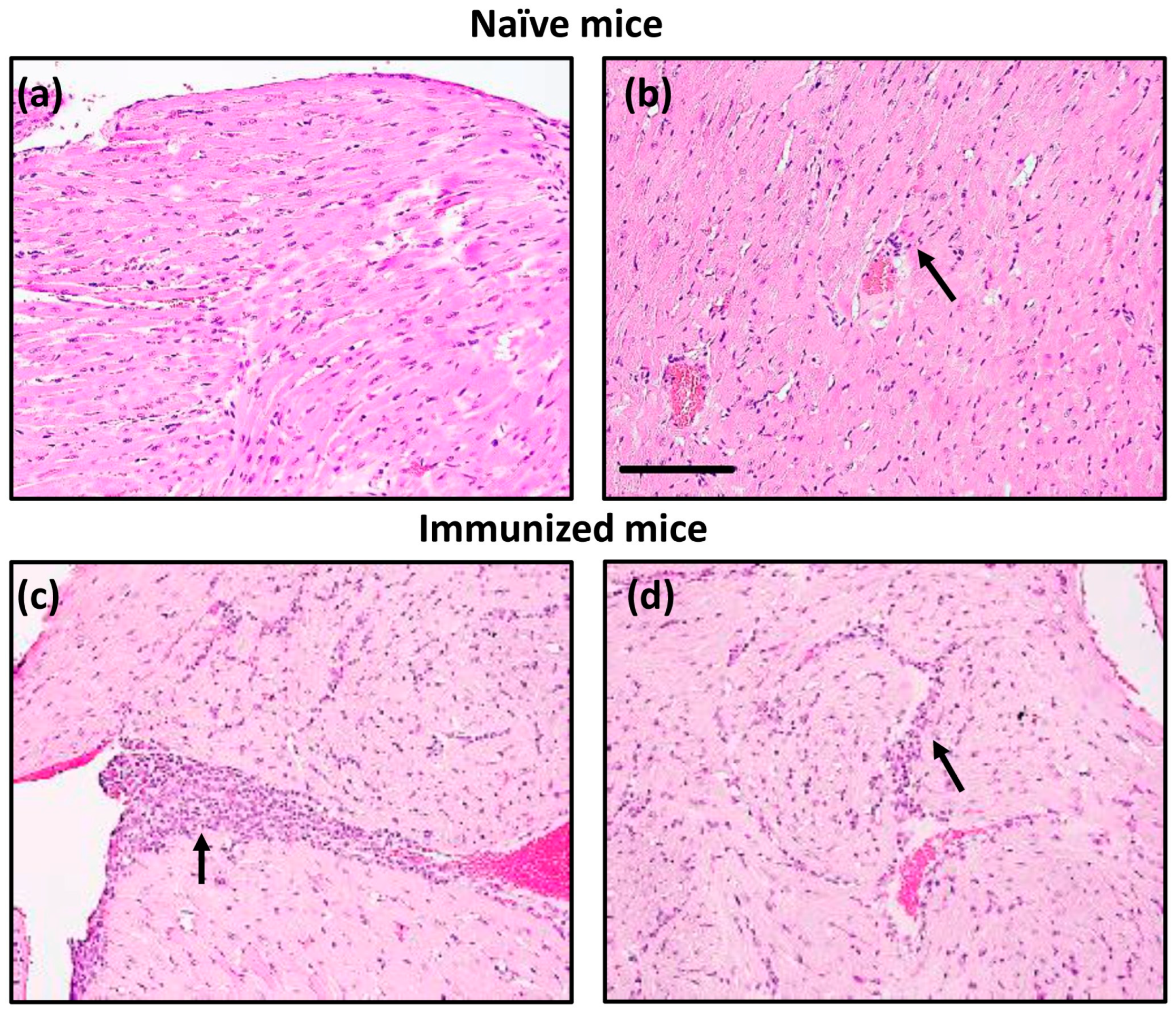{kind=link}
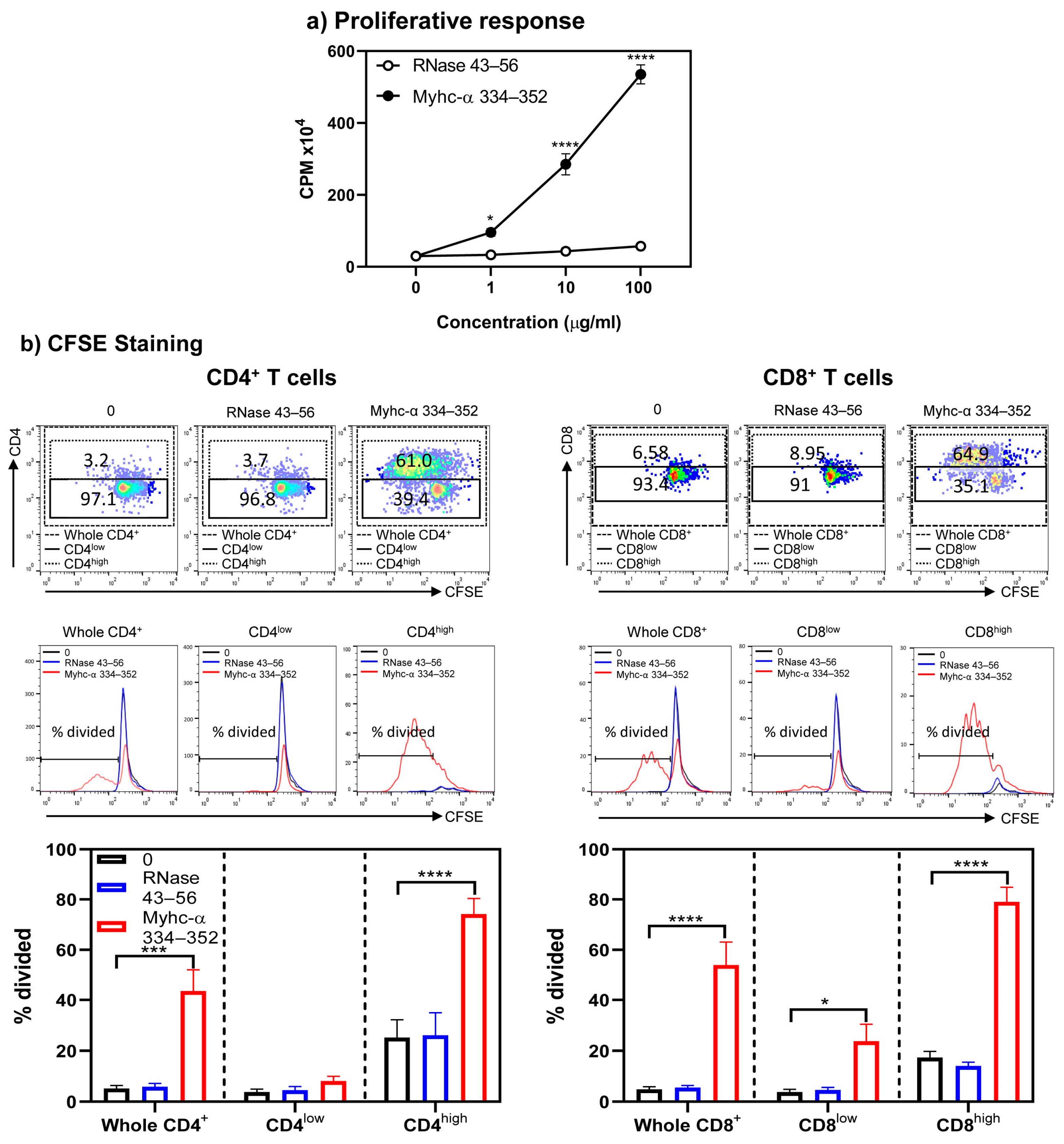{kind=link}
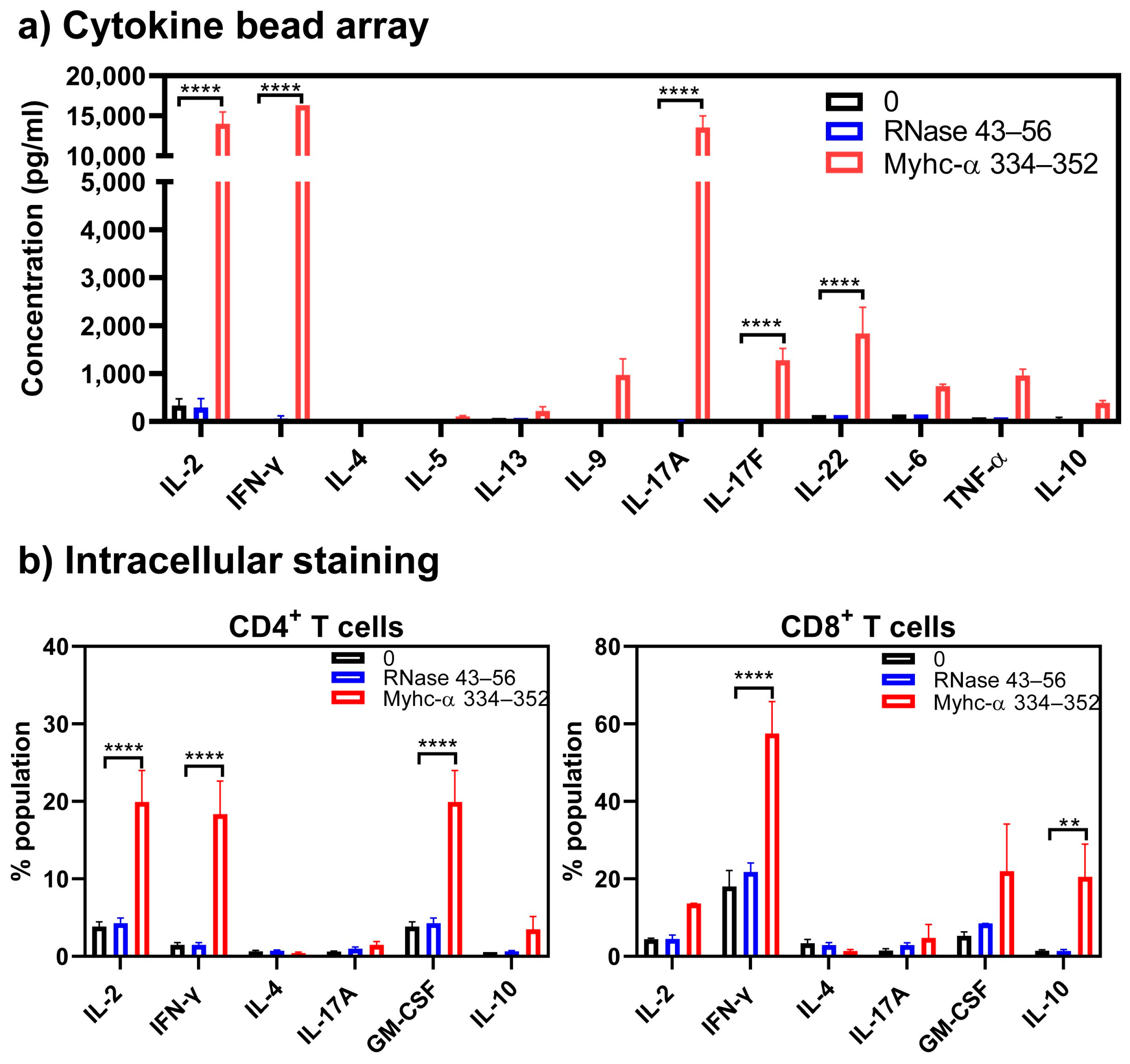{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation and Screening of TCR Transgenic (Tg) Mice
2.2. Peptide Synthesis
2.3. Immunophenotyping by Flow Cytometry
2.3.1. Surface Staining
2.3.2. Intracellular Staining
Transcription Factors
Cytokines
2.3.3. Cytotoxic T Cell (CTL) Markers
2.4. RNA Extraction and Quantitative Polymerase Chain Reaction
2.5. Proliferation Assay
2.6. Purification of Mouse Cardiac Myosin
2.7. Cytokine Bead Array Analysis
2.8. Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE) Assay
2.9. Immunization
2.10. Histopathology
2.11. Statistical Analysis
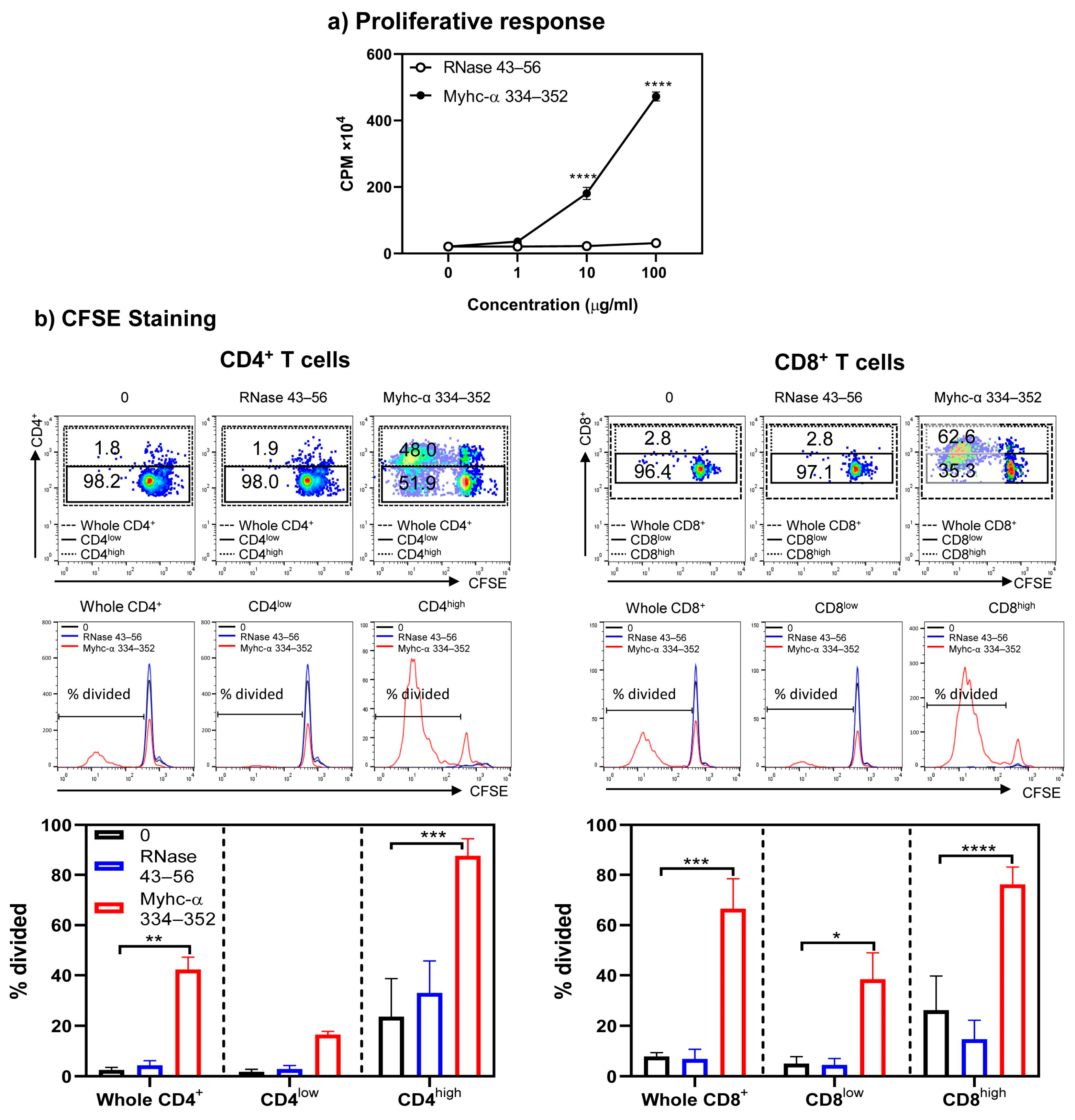
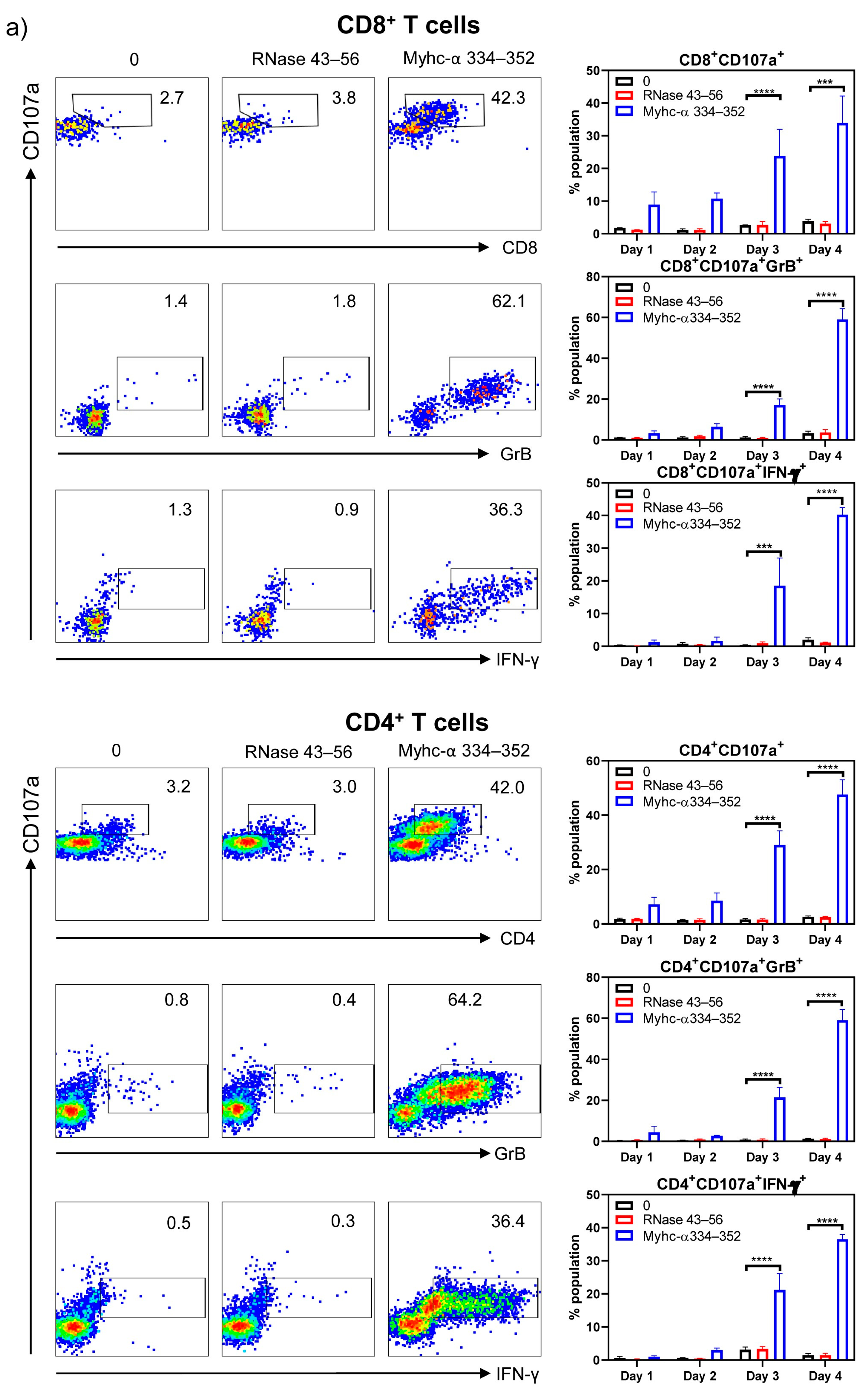
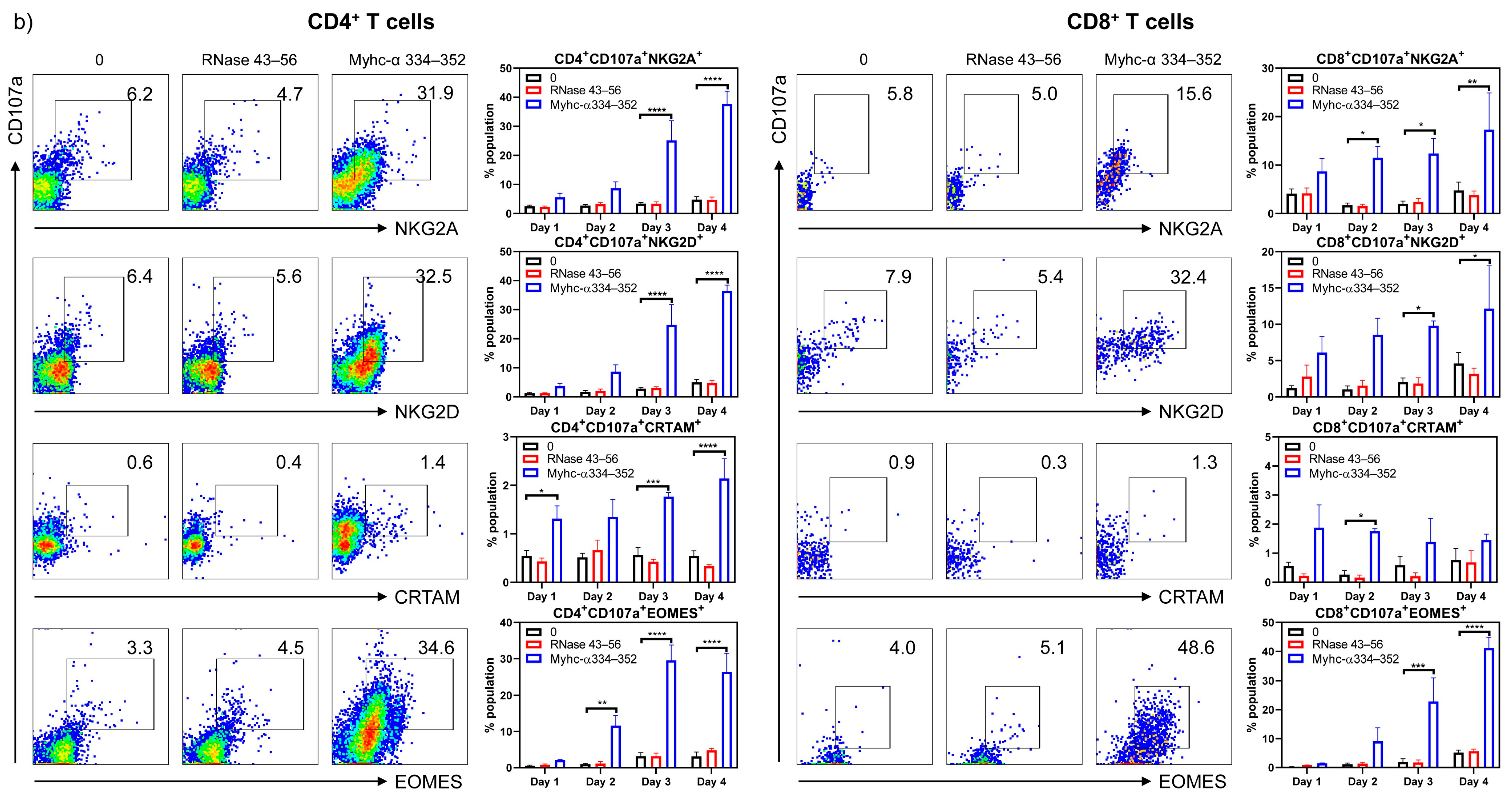
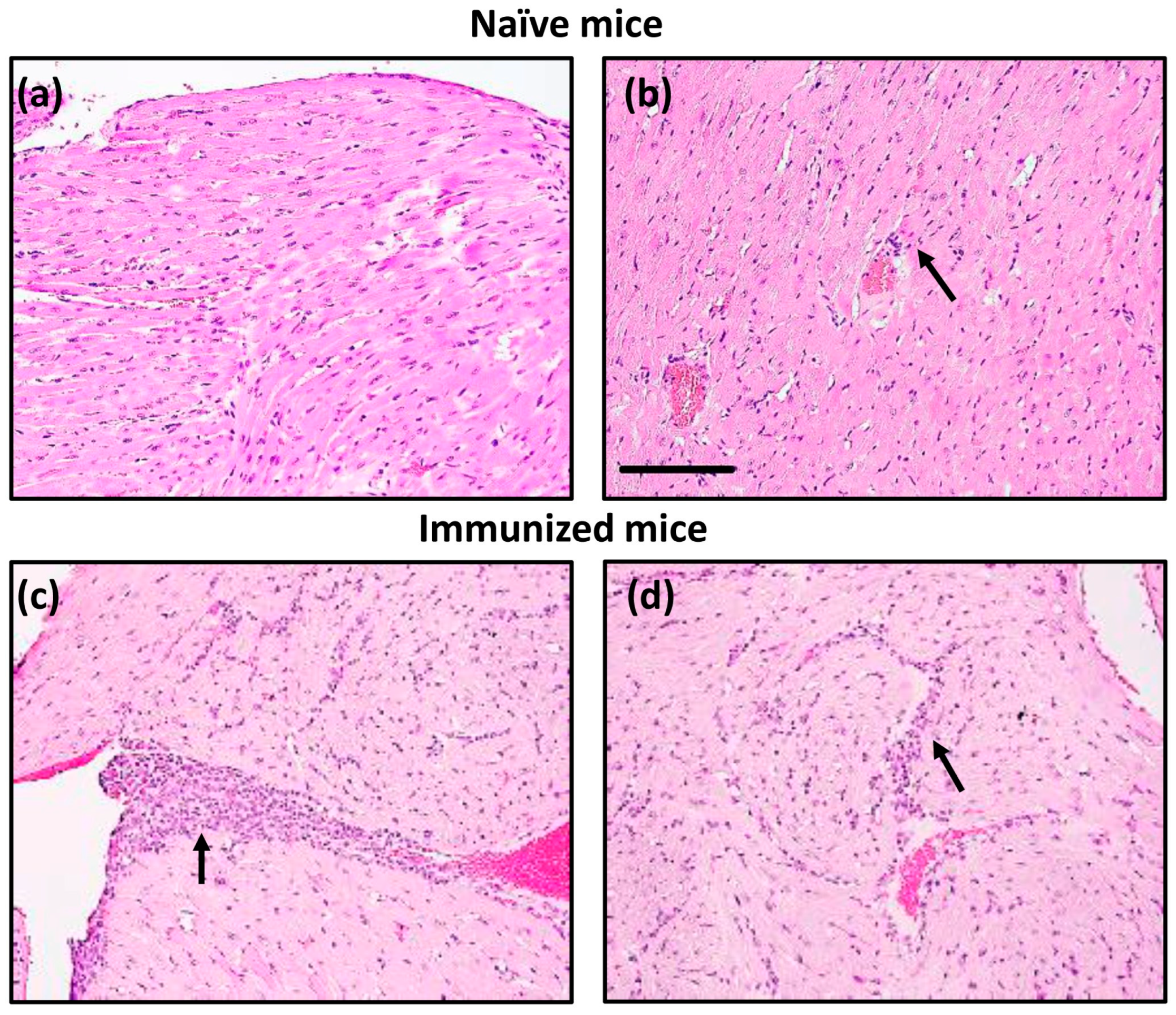
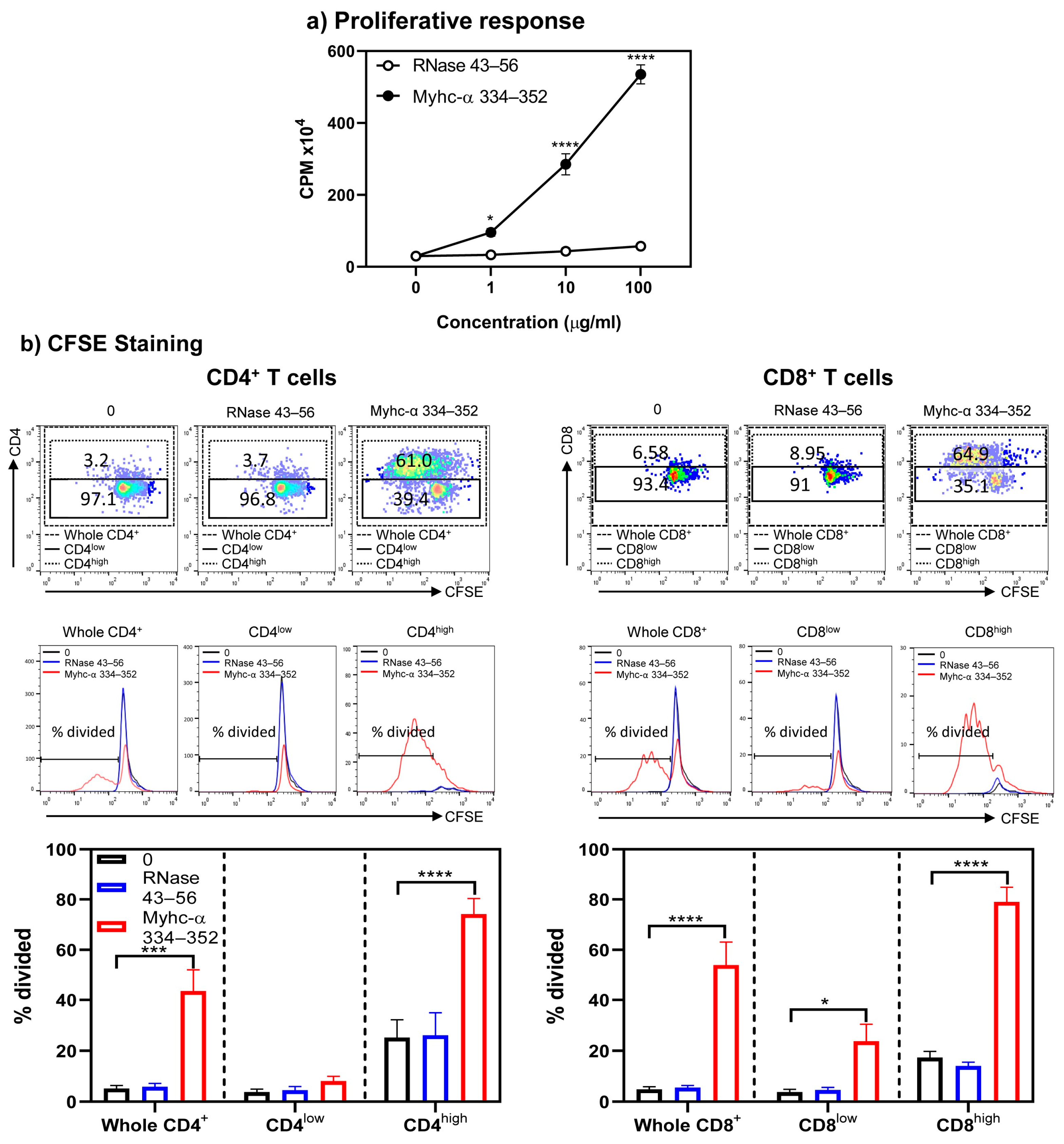
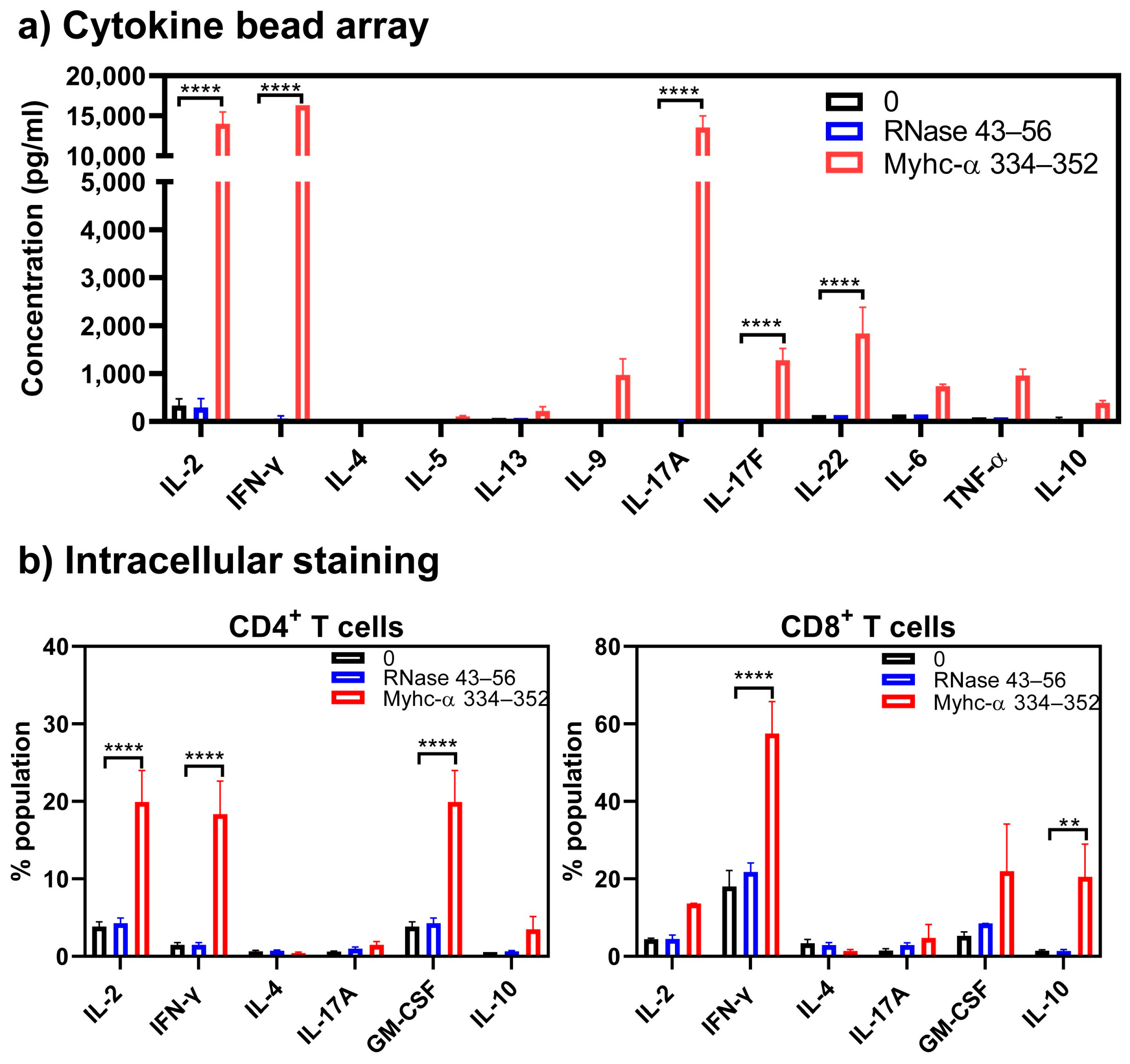
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, L.; Sun, R.; Liu, M.; Zheng, Y.; Zhang, P. The Inflammatory Heart Diseases: Causes, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 72, 851–855. [Google Scholar] [CrossRef]
- Fairweather, D.; Rose, N.R. Inflammatory heart disease: A role for cytokines. Lupus 2005, 14, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Fabre, A.; Sheppard, M.N. Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart 2006, 92, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Drory, Y.; Turetz, Y.; Hiss, Y.; Lev, B.; Fisman, E.Z.; Pines, A.; Kramer, M.R. Sudden unexpected death in persons < 40 years of age. Am. J. Cardiol. 1991, 68, 1388–1392. [Google Scholar] [PubMed]
- Golpour, A.; Patriki, D.; Hanson, P.J.; McManus, B.; Heidecker, B. Epidemiological Impact of Myocarditis. J. Clin. Med. 2021, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Won, T.; Kalinoski, H.M.; Wood, M.K.; Hughes, D.M.; Jaime, C.M.; Delgado, P.; Talor, M.V.; Lasrado, N.; Reddy, J.; Cihakova, D. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. 2022, 41, 111611. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, M.; Golden, D.L.A.; Mahmood, S.S.; Alvi, R.M.; Mercaldo, N.D.; Hassan, M.Z.O.; Banerji, D.; Rokicki, A.; Mulligan, C.; Murphy, S.P.T.; et al. Influenza vaccination and myocarditis among patients receiving immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 53. [Google Scholar] [CrossRef]
- Bozkurt, B.; Kamat, I.; Hotez, P.J. Myocarditis with COVID-19 mRNA Vaccines. Circulation 2021, 144, 471–484. [Google Scholar] [CrossRef]
- Leone, O.; Pieroni, M.; Rapezzi, C.; Olivotto, I. The spectrum of myocarditis: From pathology to the clinics. Virchows Arch. 2019, 475, 279–301. [Google Scholar] [CrossRef]
- Schultz, J.C.; Hilliard, A.A.; Cooper, L.T.; Rihal, C.S. Diagnosis and treatment of viral myocarditis. Mayo Clin. Proc. 2009, 84, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T., Jr. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Avalos, I.B. The heart in rheumatic, autoimmune and inflammatory diseases. Eur. J. Rheumatol. 2017, 4, 177. [Google Scholar] [CrossRef]
- Sur, M.; Rasquinha, M.T.; Arumugam, R.; Massilamany, C.; Gangaplara, A.; Mone, K.; Lasrado, N.; Yalaka, B.; Doiphode, A.; Gurumurthy, C.; et al. Transgenic Mice Expressing Functional TCRs Specific to Cardiac Myhc-alpha 334-352 on Both CD4 and CD8 T Cells Are Resistant to the Development of Myocarditis on C57BL/6 Genetic Background. Cells 2023, 12, 2346. [Google Scholar] [CrossRef] [PubMed]
- Massilamany, C.; Gangaplara, A.; Steffen, D.; Reddy, J. Identification of novel mimicry epitopes for cardiac myosin heavy chain-alpha that induce autoimmune myocarditis in A/J mice. Cell. Immunol. 2011, 271, 438–449. [Google Scholar] [CrossRef]
- Jia, T.; Anandhan, A.; Massilamany, C.; Rajasekaran, R.A.; Franco, R.; Reddy, J. Association of Autophagy in the Cell Death Mediated by Dihydrotestosterone in Autoreactive T Cells Independent of Antigenic Stimulation. J. Neuroimmune Pharmacol. 2015, 10, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Ashby, K.M.; Hogquist, K.A. A guide to thymic selection of T cells. Nat. Rev. Immunol. 2023. [Google Scholar] [CrossRef]
- Lasrado, N.; Arumugam, R.; Rasquinha, M.T.; Sur, M.; Steffen, D.; Reddy, J. Mt10-CVB3 Vaccine Virus Protects against CVB4 Infection by Inducing Cross-Reactive, Antigen-Specific Immune Responses. Microorganisms 2021, 9, 2323. [Google Scholar] [CrossRef]
- Quah, B.J.; Parish, C.R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 2010, 44, e2259. [Google Scholar] [CrossRef]
- Krishnan, B.; Massilamany, C.; Basavalingappa, R.H.; Gangaplara, A.; Rajasekaran, R.A.; Afzal, M.Z.; Khalilzad-Sharghi, V.; Zhou, Y.; Riethoven, J.J.; Nandi, S.S.; et al. Epitope Mapping of SERCA2a Identifies an Antigenic Determinant That Induces Mainly Atrial Myocarditis in A/J Mice. J. Immunol. 2018, 200, 523–537. [Google Scholar] [CrossRef]
- Kosmrlj, A.; Jha, A.K.; Huseby, E.S.; Kardar, M.; Chakraborty, A.K. How the thymus designs antigen-specific and self-tolerant T cell receptor sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 16671–16676. [Google Scholar] [CrossRef] [PubMed]
- Shiverick, K.T.; Thomas, L.L.; Alpert, N.R. Purification of cardiac myosin. Application to hypertrophied myocardium. Biochim. Biophys. Acta 1975, 393, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Nicholson, L.B.; Legge, K.L.; Turchin, V.; Zaghouani, H.; Kuchroo, V.K. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: Mechanisms of selection of the self-reactive repertoire. J. Exp. Med. 2000, 191, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Kuchroo, V.K. Expression of self-antigen in the thymus: A little goes a long way. J. Exp. Med. 2003, 198, 1627–1629. [Google Scholar] [CrossRef] [PubMed]
- Nindl, V.; Maier, R.; Ratering, D.; De Giuli, R.; Zust, R.; Thiel, V.; Scandella, E.; Di Padova, F.; Kopf, M.; Rudin, M.; et al. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur. J. Immunol. 2012, 42, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Havari, E.; Pinto, S.; Gottumukkala, R.V.; Cornivelli, L.; Raddassi, K.; Matsui, T.; Rosenzweig, A.; Bronson, R.T.; Smith, R.; et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J. Clin. Investig. 2011, 121, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.G.; Tyler, A.N.; Allen, P.M. T cell recognition of bovine ribonuclease. Self/non-self discrimination at the level of binding to the I-Ak molecule. J. Immunol. 1988, 141, 4124–4128. [Google Scholar] [CrossRef]
- Donermeyer, D.L.; Beisel, K.W.; Allen, P.M.; Smith, S.C. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J. Exp. Med. 1995, 182, 1291–1300. [Google Scholar] [CrossRef]
- Massilamany, C.; Gangaplara, A.; Basavalingappa, R.H.; Rajasekaran, R.A.; Khalilzad-Sharghi, V.; Han, Z.; Othman, S.; Steffen, D.; Reddy, J. Localization of CD8 T cell epitope within cardiac myosin heavy chain-alpha334-352 that induces autoimmune myocarditis in A/J mice. Int. J. Cardiol. 2016, 202, 311–321. [Google Scholar] [CrossRef]
- Massilamany, C.; Gangaplara, A.; Chapman, N.; Rose, N.; Reddy, J. Detection of cardiac myosin heavy chain-alpha-specific CD4 cells by using MHC class II/IA(k) tetramers in A/J mice. J. Immunol. Methods 2011, 372, 107–118. [Google Scholar] [CrossRef]
- Mohan, J.F.; Unanue, E.R. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat. Rev. Immunol. 2012, 12, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Carrero, J.A.; Unanue, E.R. Distinct recognition by two subsets of T cells of an MHC class II-peptide complex. Proc. Natl. Acad. Sci. USA 2002, 99, 8844–8849. [Google Scholar] [CrossRef] [PubMed]
- Lovitch, S.B.; Esparza, T.J.; Schweitzer, G.; Herzog, J.; Unanue, E.R. Activation of type B T cells after protein immunization reveals novel pathways of in vivo presentation of peptides. J. Immunol. 2007, 178, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.L.; Evavold, B.D. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2005, 35, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Chandwaskar, R.; Lee, D.H.; Sullivan, J.M.; Solomon, A.; Rodriguez-Manzanet, R.; Greve, B.; Sobel, R.A.; Kuchroo, V.K. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J. Immunol. 2012, 188, 2084–2092. [Google Scholar] [CrossRef]
- Wu, L.; Ong, S.; Talor, M.V.; Barin, J.G.; Baldeviano, G.C.; Kass, D.A.; Bedja, D.; Zhang, H.; Sheikh, A.; Margolick, J.B.; et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J. Exp. Med. 2014, 211, 1449–1464. [Google Scholar] [CrossRef]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef]
- Sonderegger, I.; Iezzi, G.; Maier, R.; Schmitz, N.; Kurrer, M.; Kopf, M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J. Exp. Med. 2008, 205, 2281–2294. [Google Scholar] [CrossRef]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef]
- Campbell, I.K.; van Nieuwenhuijze, A.; Segura, E.; O’Donnell, K.; Coghill, E.; Hommel, M.; Gerondakis, S.; Villadangos, J.A.; Wicks, I.P. Differentiation of inflammatory dendritic cells is mediated by NF-kappaB1-dependent GM-CSF production in CD4 T cells. J. Immunol. 2011, 186, 5468–5477. [Google Scholar] [CrossRef]
- Li, T.; Smith, M.; Abdussamad, M.; Katz, G.; Catalfamo, M. A flow-cytometry-based assay to assess granule exocytosis and GZB delivery by human CD8 T cells and NK cells. STAR Protoc. 2023, 4, 101939. [Google Scholar] [CrossRef]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Aktas, E.; Kucuksezer, U.C.; Bilgic, S.; Erten, G.; Deniz, G. Relationship between CD107a expression and cytotoxic activity. Cell. Immunol. 2009, 254, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.R.; Koup, R.A. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol. 2004, 75, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.M.; Christensen, J.R.; Thomas, D.B. Differential induction of CD94 and NKG2 in CD4 helper T cells. A consequence of influenza virus infection and interferon-gamma? Immunology 2007, 121, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Saez-Borderias, A.; Guma, M.; Angulo, A.; Bellosillo, B.; Pende, D.; Lopez-Botet, M. Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur. J. Immunol. 2006, 36, 3198–3206. [Google Scholar] [CrossRef]
- Takeuchi, A.; Badr Mel, S.; Miyauchi, K.; Ishihara, C.; Onishi, R.; Guo, Z.; Sasaki, Y.; Ike, H.; Takumi, A.; Tsuji, N.M.; et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 2016, 213, 123–138. [Google Scholar] [CrossRef]
- Dejean, A.S.; Joulia, E.; Walzer, T. The role of Eomes in human CD4 T cell differentiation: A question of context. Eur. J. Immunol. 2019, 49, 38–41. [Google Scholar] [CrossRef]
- Qui, H.Z.; Hagymasi, A.T.; Bandyopadhyay, S.; St Rose, M.C.; Ramanarasimhaiah, R.; Menoret, A.; Mittler, R.S.; Gordon, S.M.; Reiner, S.L.; Vella, A.T.; et al. CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J. Immunol. 2011, 187, 3555–3564. [Google Scholar] [CrossRef]
- Pradella, F.; Boldrini, V.O.; Marques, A.M.; Morais, G.A.D.; Francelin, C.; Cocenza, R.S.; Lima, V.C.; Maurilio Bonora, J.; Brunetti, N.S.; Campos, B.B.; et al. Cytotoxic Activity of CD4 T Cells During the Early Stage of Autoimmune Neuroinflammation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Meyers, J.H.; Ryu, A.; Monney, L.; Nguyen, K.; Greenfield, E.A.; Freeman, G.J.; Kuchroo, V.K. Cutting edge: CD94/NKG2 is expressed on Th1 but not Th2 cells and costimulates Th1 effector functions. J. Immunol. 2002, 169, 5382–5386. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat-Zucman, S.; Paul, P.; Gornet, J.M.; Douay, C.; et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef] [PubMed]
- Ruck, T.; Bittner, S.; Gross, C.C.; Breuer, J.; Albrecht, S.; Korr, S.; Gobel, K.; Pankratz, S.; Henschel, C.M.; Schwab, N.; et al. CD4+NKG2D+ T cells exhibit enhanced migratory and encephalitogenic properties in neuroinflammation. PLoS ONE 2013, 8, e81455. [Google Scholar] [CrossRef]
- Yeh, J.H.; Sidhu, S.S.; Chan, A.C. Regulation of a late phase of T cell polarity and effector functions by Crtam. Cell 2008, 132, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhu, Z.; Hu, J.; Sun, J.; Wo, Y.; Wang, X.; Zou, H.; Li, B.; Zhang, Y. Downregulated cytotoxic CD8(+) T-cell identifies with the NKG2A-soluble HLA-E axis as a predictive biomarker and potential therapeutic target in keloids. Cell Mol. Immunol. 2022, 19, 527–539. [Google Scholar] [CrossRef]
- Chen, Y.; Xin, Z.; Huang, L.; Zhao, L.; Wang, S.; Cheng, J.; Wu, P.; Chai, Y. CD8(+) T Cells Form the Predominant Subset of NKG2A(+) Cells in Human Lung Cancer. Front. Immunol. 2019, 10, 3002. [Google Scholar] [CrossRef]
- Prajapati, K.; Perez, C.; Rojas, L.B.P.; Burke, B.; Guevara-Patino, J.A. Functions of NKG2D in CD8(+) T cells: An opportunity for immunotherapy. Cell Mol. Immunol. 2018, 15, 470–479. [Google Scholar] [CrossRef]
- Takeuchi, A.; Itoh, Y.; Takumi, A.; Ishihara, C.; Arase, N.; Yokosuka, T.; Koseki, H.; Yamasaki, S.; Takai, Y.; Miyoshi, J.; et al. CRTAM confers late-stage activation of CD8+ T cells to regulate retention within lymph node. J. Immunol. 2009, 183, 4220–4228. [Google Scholar] [CrossRef]
- Medina-Contreras, O.; Soldevila, G.; Patino-Lopez, G.; Canche-Pool, E.; Valle-Rios, R.; Ortiz-Navarrete, V. Role of CRTAM during mouse early T lymphocytes development. Dev. Comp. Immunol. 2010, 34, 196–202. [Google Scholar] [CrossRef]
- Pearce, E.L.; Mullen, A.C.; Martins, G.A.; Krawczyk, C.M.; Hutchins, A.S.; Zediak, V.P.; Banica, M.; DiCioccio, C.B.; Gross, D.A.; Mao, C.A.; et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 2003, 302, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.M.; Cooper, L.T.; Kem, D.C.; Stavrakis, S.; Kosanke, S.D.; Shevach, E.M.; Fairweather, D.; Stoner, J.A.; Cox, C.J.; Cunningham, M.W. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 2016, 1, e85851. [Google Scholar] [CrossRef] [PubMed]
- Peeters, L.M.; Vanheusden, M.; Somers, V.; Van Wijmeersch, B.; Stinissen, P.; Broux, B.; Hellings, N. Cytotoxic CD4+ T Cells Drive Multiple Sclerosis Progression. Front. Immunol. 2017, 8, 1160. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Meng, S.; Tang, D.; Wang, T.; Ding, L.; Yu, H.; Li, H.; Liu, D.; Dai, Y.; Yang, M. Single-Cell RNA Sequencing Reveals the Expansion of Cytotoxic CD4(+) T Lymphocytes and a Landscape of Immune Cells in Primary Sjogren’s Syndrome. Front. Immunol. 2020, 11, 594658. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Tian, Z.; Zhang, M.; Yang, W.; Tang, J.; Wu, Y.; Ni, B. NKG2D(+)CD4(+) T Cells Kill Regulatory T Cells in a NKG2D-NKG2D Ligand- Dependent Manner in Systemic Lupus Erythematosus. Sci. Rep. 2017, 7, 1288. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, T.; Wagner, U.G.; Goronzy, J.J.; Weyand, C.M. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998, 41, 2108–2116. [Google Scholar] [CrossRef] [PubMed]
- Cenerenti, M.; Saillard, M.; Romero, P.; Jandus, C. The Era of Cytotoxic CD4 T Cells. Front. Immunol. 2022, 13, 867189. [Google Scholar] [CrossRef]
- Grzechocinska, J.; Tyminska, A.; Giordani, A.S.; Wysinska, J.; Ostrowska, E.; Baritussio, A.; Caforio, A.L.P.; Grabowski, M.; Marcolongo, R.; Ozieranski, K. Immunosuppressive Therapy of Biopsy-Proven, Virus-Negative, Autoimmune/Immune-Mediated Myocarditis-Focus on Azathioprine: A Review of Existing Evidence and Future Perspectives. Biology 2023, 12, 356. [Google Scholar] [CrossRef]
- Escher, F.; Kuhl, U.; Lassner, D.; Poller, W.; Westermann, D.; Pieske, B.; Tschope, C.; Schultheiss, H.P. Long-term outcome of patients with virus-negative chronic myocarditis or inflammatory cardiomyopathy after immunosuppressive therapy. Clin. Res. Cardiol. 2016, 105, 1011–1020. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sur, M.; Rasquinha, M.T.; Mone, K.; Massilamany, C.; Lasrado, N.; Gurumurthy, C.; Sobel, R.A.; Reddy, J. Investigation into Cardiac Myhc-α 334–352-Specific TCR Transgenic Mice Reveals a Role for Cytotoxic CD4 T Cells in the Development of Cardiac Autoimmunity. Cells 2024, 13, 234. https://doi.org/10.3390/cells13030234
Sur M, Rasquinha MT, Mone K, Massilamany C, Lasrado N, Gurumurthy C, Sobel RA, Reddy J. Investigation into Cardiac Myhc-α 334–352-Specific TCR Transgenic Mice Reveals a Role for Cytotoxic CD4 T Cells in the Development of Cardiac Autoimmunity. Cells. 2024; 13(3):234. https://doi.org/10.3390/cells13030234
Chicago/Turabian StyleSur, Meghna, Mahima T. Rasquinha, Kiruthiga Mone, Chandirasegaran Massilamany, Ninaad Lasrado, Channabasavaiah Gurumurthy, Raymond A. Sobel, and Jay Reddy. 2024. "Investigation into Cardiac Myhc-α 334–352-Specific TCR Transgenic Mice Reveals a Role for Cytotoxic CD4 T Cells in the Development of Cardiac Autoimmunity" Cells 13, no. 3: 234. https://doi.org/10.3390/cells13030234
APA StyleSur, M., Rasquinha, M. T., Mone, K., Massilamany, C., Lasrado, N., Gurumurthy, C., Sobel, R. A., & Reddy, J. (2024). Investigation into Cardiac Myhc-α 334–352-Specific TCR Transgenic Mice Reveals a Role for Cytotoxic CD4 T Cells in the Development of Cardiac Autoimmunity. Cells, 13(3), 234. https://doi.org/10.3390/cells13030234