
Emerging Roles of Cullin-RING Ubiquitin Ligases in Cardiac Development

 , , and
, , and 
Abstract
1. Introduction
2. Neddylation of Cullins in Cardiac Development
3. Deneddylation of Cullins in Cardiac Development
4. RBX1 and RBX2
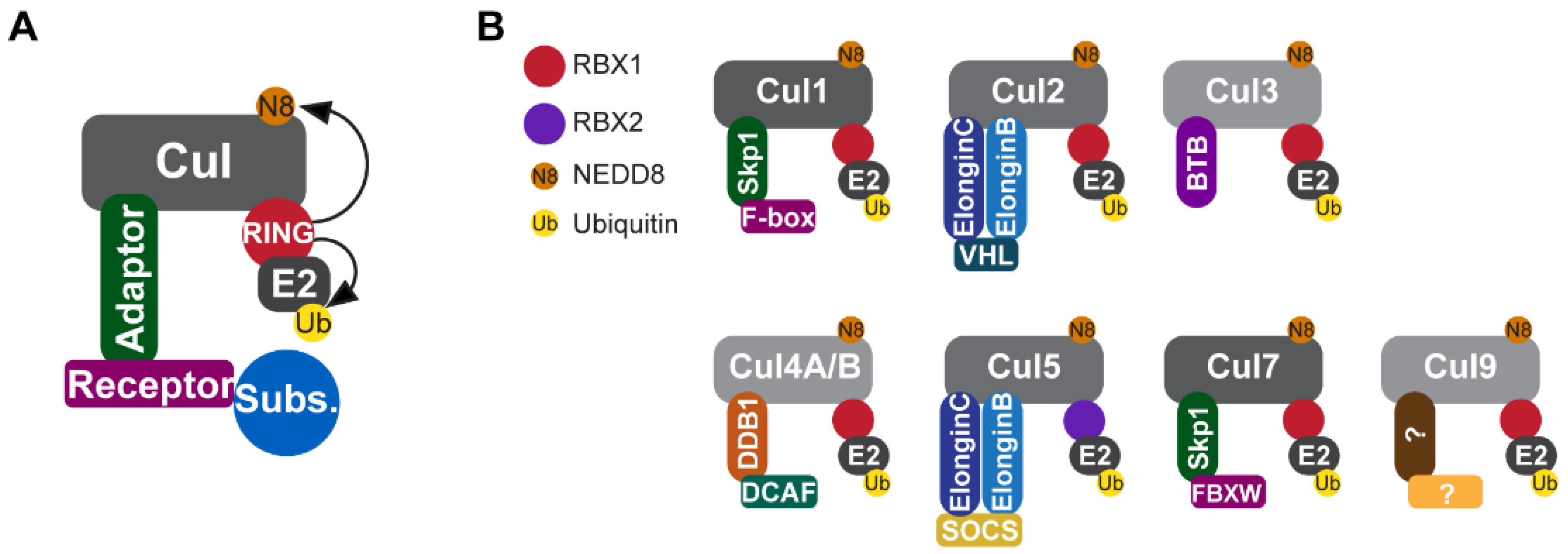
5. Cullin-RING Ubiquitin Ligases
5.1. CRL1
5.2. CRL2
5.3. CRL3
5.4. CRL4A/B
5.5. CRL5
5.6. CRL7 and CRL9
6. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rufaihah, A.J.; Chen, C.K.; Yap, C.H.; Mattar, C.N.Z. Mending a broken heart: In vitro, in vivo and in silico models of congenital heart disease. Dis. Model. Mech. 2021, 14, dmm047522. [Google Scholar] [CrossRef] [PubMed]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.L. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- van der Linde, D.; Konings, E.E.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Van der Bom, T.; Zomer, A.C.; Zwinderman, A.H.; Meijboom, F.J.; Bouma, B.J.; Mulder, B.J. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2011, 8, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Später, D.; Hansson, E.M.; Zangi, L.; Chien, K.R. How to make a cardiomyocyte. Development 2014, 141, 4418–4431. [Google Scholar] [CrossRef]
- Waardenberg, A.J.; Ramialison, M.; Bouveret, R.; Harvey, R.P. Genetic networks governing heart development. Cold Spring Harb. Perspect. Med. 2014, 4, a013839. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef]
- Meilhac, S.M.; Lescroart, F.; Blanpain, C.; Buckingham, M.E. Cardiac cell lineages that form the heart. Cold Spring Harb. Perspect. Med. 2014, 4, a013888. [Google Scholar] [CrossRef]
- Villavicencio-Guzmán, L.; Sánchez-Gómez, C.; Jaime-Cruz, R.; Ramírez-Fuentes, T.C.; Patiño-Morales, C.C.; Salazar-García, M. Human Heart Morphogenesis: A New Vision Based on In Vivo Labeling and Cell Tracking. Life 2023, 13, 165. [Google Scholar] [CrossRef]
- Miquerol, L.; Kelly, R.G. Organogenesis of the vertebrate heart. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 17–29. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, H.; Qu, X.; Chang, C.P.; Shou, W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am. J. Med. Genet. Part C Semin. Med. Genet. 2013, 163C, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Grego-Bessa, J.; Gómez-Apiñaniz, P.; Prados, B.; Gómez, M.J.; MacGrogan, D.; de la Pompa, J.L. NRG1 Regulates Cardiomyocyte Migration and Cell Cycle in Ventricular Development. Circ. Res. 2023, 133, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Miao, L.J.; Shieh, D.; Spiotto, E.; Li, J.; Zhou, B.; Paul, A.; Schwartz, R.J.; Firulli, A.B.; Singer, H.A.; et al. Single-Cell Lineage Tracing Reveals That Oriented Cell Division Contributes to Trabecular Morphogenesis and Regional Specification. Cell Rep. 2016, 15, 158–170. [Google Scholar] [CrossRef]
- Liu, J.D.; Bressan, M.; Hassel, D.; Huisken, J.; Staudt, D.; Kikuchi, K.; Poss, K.D.; Mikawa, T.; Stainier, D.Y.R. A dual role for ErbB2 signaling in cardiac trabeculation. Development 2010, 137, 3867–3875. [Google Scholar] [CrossRef]
- Miao, L.J.; Li, J.J.; Li, J.; Lu, Y.Y.; Shieh, D.; Mazurkiewicz, J.E.; Barroso, M.; Schwarz, J.J.; Xin, H.B.; Singer, H.A.; et al. Cardiomyocyte orientation modulated by the Numb family proteins-N-cadherin axis is essential for ventricular wall morphogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 15560–15569. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Singh, B.N.; Yucel, D.; Garay, B.I.; Tolkacheva, E.G.; Kyba, M.; Perlingeiro, R.C.R.; Berlo, J.; Ogle, B.M. Proliferation and Maturation: Janus and the Art of Cardiac Tissue Engineering. Circ. Res. 2023, 132, 519–540. [Google Scholar] [CrossRef]
- Salameh, S.; Ogueri, V.; Posnack, N.G. Adapting to a new environment: Postnatal maturation of the human cardiomyocyte. J. Physiol. 2023, 601, 2593–2619. [Google Scholar] [CrossRef] [PubMed]
- Beisaw, A.; Wu, C.C. Cardiomyocyte maturation and its reversal during cardiac regeneration. Dev. Dyn. 2024, 253, 8–27. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Consortium, G.T.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e19. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.; Keung, W.; Liang, Y.; Ramalingam, R.; Yan, B.; Zhang, S.; Chopra, A.; Moore, J.; Herren, A.; Lieu, D.K.; et al. Proteomic Analysis of Human Pluripotent Stem Cell-Derived, Fetal, and Adult Ventricular Cardiomyocytes Reveals Pathways Crucial for Cardiac Metabolism and Maturation. Circ. Cardiovasc. Genet. 2015, 8, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin proteasome pathway on protein death and cell life. EMBO 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Cui, D.; Xiong, X.; Zhao, Y. The characteristics and roles of beta-TrCP1/2 in carcinogenesis. FEBS J. 2021, 288, 3351–3374. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Sun, Y. Cullin-RING Ligases and Protein Neddylation Biology and Therapeutics; Sun, Y., Wei, W., Jin, J., Eds.; Springer: Singapore, 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr. Pharm. Des. 2013, 19, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef]
- Harper, J.W.; Schulman, B.A. Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis. Annu. Rev. Biochem. 2021, 90, 403–429. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, H.; Wu, B.; You, S.; Wu, S.; Lu, S.; Wang, P.; Cao, L.; Zhang, N.; Sun, Y. E3 Ubiquitin ligase NEDD4 family-regulatory network in cardiovascular disease. Int. J. Biol. Sci. 2020, 16, 2727–2740. [Google Scholar] [CrossRef]
- Jang, S.M.; Redon, C.E.; Thakur, B.L.; Bahta, M.K.; Aladjem, M.I. Regulation of cell cycle drivers by Cullin-RING ubiquitin ligases. Exp. Mol. Med. 2020, 52, 1637–1651. [Google Scholar] [CrossRef]
- Walden, H.; Podgorski, M.S.; Huang, D.T.; Miller, D.W.; Howard, R.J.; Minor, D.L., Jr.; Holton, J.M.; Schulman, B.A. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 2003, 12, 1427–1437. [Google Scholar] [CrossRef]
- Zou, J.; Ma, W.; Li, J.; Littlejohn, R.; Zhou, H.; Kim, I.M.; Fulton, D.J.R.; Chen, W.; Weintraub, N.L.; Zhou, J.; et al. Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4101–E4110. [Google Scholar] [CrossRef]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (YAP1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lei, W.; Xiang, X.; Zhang, L.; Lei, J.; Gong, Y.; Song, M.; Wang, Y.; Fang, Z.; Yu, F.; et al. CULLIN 4A (CUL4A), a direct target of miR-9 and miR-137, promotes gastric cancer proliferation and invasion by regulating the Hippo signaling pathway. Oncotarget 2016, 7, 10037–10050. [Google Scholar] [CrossRef]
- Zou, J.; Ma, W.; Littlejohn, R.; Li, J.; Stansfield, B.K.; Kim, I.M.; Liu, J.; Zhou, J.; Weintraub, N.L.; Su, H. Transient inhibition of neddylation at neonatal stage evokes reversible cardiomyopathy and predisposes the heart to isoproterenol-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1406–H1416. [Google Scholar] [CrossRef]
- Zou, J.; Wang, W.; Lu, Y.; Ayala, J.; Dong, K.; Zhou, H.; Wang, J.; Chen, W.; Weintraub, N.L.; Zhou, J.; et al. Neddylation is required for perinatal cardiac development through stimulation of metabolic maturation. Cell Rep. 2023, 42, 112018. [Google Scholar] [CrossRef]
- Guimaraes-Camboa, N.; Stowe, J.; Aneas, I.; Sakabe, N.; Cattaneo, P.; Henderson, L.; Kilberg, M.S.; Johnson, R.S.; Chen, J.; McCulloch, A.D.; et al. HIF1alpha Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev. Cell 2015, 33, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hickey, R.P.; Yeh, J.L.; Liu, D.; Dadak, A.; Young, L.H.; Johnson, R.S.; Giordano, F.J. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004, 18, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Curtis, V.F.; Ehrentraut, S.F.; Campbell, E.L.; Glover, L.E.; Bayless, A.; Kelly, C.J.; Kominsky, D.J.; Colgan, S.P. Stabilization of HIF through inhibition of CULLIN-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2014, 29, 208–215. [Google Scholar] [CrossRef]
- Ryu, J.H.; Li, S.H.; Park, H.S.; Park, J.W.; Lee, B.; Chun, Y.S. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J. Biol. Chem. 2011, 286, 6963–6970. [Google Scholar] [CrossRef]
- Heir, P.; Sufan, R.I.; Greer, S.N.; Poon, B.P.; Lee, J.E.; Ohh, M. DCNL1 functions as a substrate sensor and activator of cullin 2-RING ligase. Mol. Cell. Biol. 2013, 33, 1621–1631. [Google Scholar] [CrossRef]
- Su, H.; Li, J.; Menon, S.; Liu, J.; Kumarapeli, A.R.; Wei, N.; Wang, X. Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ. Res. 2011, 108, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Li, J.; Osinska, H.; Li, F.; Robbins, J.; Liu, J.; Wei, N.; Wang, X. The COP9 signalosome is required for autophagy, proteasome-mediated proteolysis, and cardiomyocyte survival in adult mice. Circ. Heart Fail. 2013, 6, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Serino, G.; Deng, X.W. The COP9 signalosome: More than a protease. Trends Biochem. Sci. 2008, 33, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, K.; Schaefer, L.; Menon, S.; Deng, X.W.; Miller, J.B.; Wei, N. Disruption of the COP9 signalosome CSN2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol. Cell Biol. 2003, 23, 6790–6797. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Walz, K.; Nakamura, H.; Carattini-Rivera, S.; Zhao, Q.; Vogel, H.; Wei, N.; Justice, M.J.; Bradley, A.; Lupski, J.R. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol. Cell Biol. 2003, 23, 6798–6808. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, K.; Yoneda-Kato, N.; Fukumoto, A.; Yamanaka, S.; Kato, J.Y. Multiple functions of JAB1 are required for early embryonic development and growth potential in mice. J. Biol. Chem. 2004, 279, 43013–43018. [Google Scholar] [CrossRef]
- Zhao, R.; Yeung, S.C.; Chen, J.; Iwakuma, T.; Su, C.H.; Chen, B.; Qu, C.; Zhang, F.; Chen, Y.T.; Lin, Y.L.; et al. Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J. Clin. Investig. 2011, 121, 851–865. [Google Scholar] [CrossRef]
- Menon, S.; Chi, H.; Zhang, H.; Deng, X.W.; Flavell, R.A.; Wei, N. COP9 signalosome subunit 8 is essential for peripheral T cell homeostasis and antigen receptor-induced entry into the cell cycle from quiescence. Nat. Immunol. 2007, 8, 1236–1245. [Google Scholar] [CrossRef]
- Su, H.; Li, F.; Ranek, M.J.; Wei, N.; Wang, X. COP9 signalosome regulates autophagosome maturation. Circulation 2011, 124, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Holt, M.R.; Mankoo, B.S.; Gautel, M. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev. Biol. 2011, 351, 46–61. [Google Scholar] [CrossRef]
- Mei, Z.L.; Wang, H.B.; Hu, Y.H.; Xiong, L. CSN6 aggravates Ang II-induced cardiomyocyte hypertrophy via inhibiting SIRT2. Exp. Cell Res. 2020, 396, 112245. [Google Scholar] [CrossRef]
- Liang, Y.; Lyon, R.C.; Pellman, J.; Bradford, W.H.; Lange, S.; Bogomolovas, J.; Dalton, N.D.; Gu, Y.; Bobar, M.; Lee, M.H.; et al. Desmosomal COP9 regulates proteome degradation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Clin. Investig. 2021, 131, e137689. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zou, J.; Littlejohn, R.; Liu, J.; Su, H. Neddylation, an Emerging Mechanism Regulating Cardiac Development and Function. Front. Physiol. 2020, 11, 612927. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Sun, Y. RBX1/ROC1-SCF E3 ubiquitin ligase is required for mouse embryogenesis and cancer cell survival. Cell Div. 2009, 4, 16. [Google Scholar] [CrossRef]
- Zhou, W.; Wei, W.; Sun, Y. Genetically engineered mouse models for functional studies of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases. Cell Res. 2013, 23, 599–619. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Davis, S.W.; Saunders, T.L.; Zhu, Y.; Sun, Y. RBX1/ROC1 disruption results in early embryonic lethality due to proliferation failure, partially rescued by simultaneous loss of p27. Proc. Natl. Acad. Sci. USA 2009, 106, 6203–6208. [Google Scholar] [CrossRef]
- Sarvari, P.; Rasouli, S.J.; Allanki, S.; Stone, O.A.; Sokol, A.M.; Graumann, J.; Stainier, D.Y.R. The E3 ubiquitin-protein ligase RBX1 regulates cardiac wall morphogenesis in zebrafish. Dev. Biol. 2021, 480, 1–12. [Google Scholar] [CrossRef]
- Yang, G.Y.; Pang, L.; Ge, H.L.; Tan, M.; Ye, W.; Liu, X.H.; Huang, F.P.; Wu, D.C.; Che, X.M.; Song, Y.; et al. Attenuation of ischemia-induced mouse brain injury by SAG, a redox-inducible antioxidant protein. J. Cereb. Blood Flow Metab. 2001, 21, 722–733. [Google Scholar] [CrossRef]
- Tan, M.; Zhao, Y.; Kim, S.J.; Liu, M.; Jia, L.; Saunders, T.L.; Zhu, Y.; Sun, Y. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev. Cell 2011, 21, 1062–1076. [Google Scholar] [CrossRef]
- Tan, M.; Li, H.; Sun, Y. Endothelial deletion of SAG/RBX2/ROC2 E3 ubiquitin ligase causes embryonic lethality and blocks tumor angiogenesis. Oncogene 2014, 33, 5211–5220. [Google Scholar] [CrossRef]
- Asmamaw, M.D.; Liu, Y.; Zheng, Y.C.; Shi, X.J.; Liu, H.M. SKP2 in the ubiquitin-proteasome system: A comprehensive review. Med. Res. Rev. 2020, 40, 1920–1949. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 2014, 14, 233–247. [Google Scholar] [CrossRef]
- Dealy, M.J.; Nguyen, K.V.T.; Lo, J.; Gstaiger, M.; Krek, W.; Elson, D.; Arbeit, J.; Kipreos, E.T.; Johnson, R.S. Loss of Cul1 results in early embryonic lethality and dysregulation of cyclin E. Nature 1999, 23, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, L.; Goodyer, W.; Kort, E.J.; Buikema, J.W.; Xu, A.; Wu, J.C.; Jovinge, S.; Wu, S.M. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development 2019, 146, dev173476. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, J.; Biju, A.; Lange, S. The Role of Cullin-RING Ligases in Striated Muscle Development, Function, and Disease. Int. J. Mol. Sci. 2020, 21, 7936. [Google Scholar] [CrossRef]
- Tetzlaff, M.T.; Yu, W.; Li, M.; Zhang, P.; Finegold, M.; Mahon, K.; Harper, J.W.; Schwartz, R.J.; Elledge, S.J. Defective cardiovascular development and elevated Cyclin E and NOTCH proteins in mice lacking the FBW7 F-box protein. Proc. Natl. Acad. Sci. USA 2004, 101, 3338–3345. [Google Scholar] [CrossRef]
- Matsumoto, A.; Onoyama, I.; Sunabori, T.; Kageyama, R.; Okano, H.; Nakayama, K.I. FBXW7-dependent degradation of NOTCH is required for control of “stemness” and neuronal-glial differentiation in neural stem cells. J. Biol. Chem. 2011, 286, 13754–13764. [Google Scholar] [CrossRef] [PubMed]
- Buhler, A.; Kustermann, M.; Bummer, T.; Rottbauer, W.; Sandri, M.; Just, S. Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish. Int. J. Mol. Sci. 2016, 17, 187. [Google Scholar] [CrossRef]
- Zaglia, T.; Milan, G.; Ruhs, A.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Carpi, A.; Carullo, P.; Pesce, P.; Sacerdoti, D.; et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Investig. 2014, 124, 2410–2424. [Google Scholar] [CrossRef]
- Jeong, H.S.; Jung, E.S.; Sim, Y.J.; Kim, S.J.; Jang, J.W.; Hong, K.S.; Lee, W.Y.; Chung, H.M.; Park, K.T.; Jung, Y.S.; et al. FBXO25 controls TBX5 and NKX2-5 transcriptional activity to regulate cardiomyocyte development. Biochim. Biophys. Acta 2015, 1849, 709–721. [Google Scholar] [CrossRef]
- Bruneau, B.G. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb. Perspect. Biol. 2013, 5, a008292. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Maher, E.R. The VHL tumour-suppressor gene paradigm. Trends Genet. 1998, 14, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Yang, H. The structure and regulation of CULLIN 2 based E3 ubiquitin ligases and their biological functions. Cell Div. 2016, 11, 7. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Patterson, A.J.; Zhang, L. Hypoxia and fetal heart development. Curr. Mol. Med. 2010, 10, 653–666. [Google Scholar] [CrossRef]
- Menendez-Montes, I.; Escobar, B.; Palacios, B.; Gomez, M.J.; Izquierdo-Garcia, J.L.; Flores, L.; Jimenez-Borreguero, L.J.; Aragones, J.; Ruiz-Cabello, J.; Torres, M.; et al. Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Dev. Cell 2016, 39, 724–739. [Google Scholar] [CrossRef]
- Knutson, A.K.; Williams, A.L.; Boisvert, W.A.; Shohet, R.V. HIF in the heart: Development, metabolism, ischemia, and atherosclerosis. J. Clin. Investig. 2021, 131, e137557. [Google Scholar] [CrossRef]
- Neary, M.T.; Ng, K.E.; Ludtmann, M.H.; Hall, A.R.; Piotrowska, I.; Ong, S.B.; Hausenloy, D.J.; Mohun, T.J.; Abramov, A.Y.; Breckenridge, R.A. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell. Cardiol. 2014, 74, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, S.; Asahi, M. Phospholamban Is Downregulated by pVHL-Mediated Degradation through Oxidative Stress in Failing Heart. Int. J. Mol. Sci. 2017, 18, 2232. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Ferdaus, M.Z.; Miller, L.N.; Agbor, L.N.; Saritas, T.; Singer, J.D.; Sigmund, C.D.; McCormick, J.A. Mutant Cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight 2017, 2, e96700. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, J.H.; Ahn, Y.H.; Kang, H.G.; Ha, I.S.; Cheong, H.I. Gordon syndrome caused by a CUL3 mutation in a patient with short stature in Korea: A case report. J. Pediatr. Endocrinol. Metab. 2022, 35, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Turer, E.; Li, X.; Zhan, X.; Choi, M.; Tang, M.; Press, A.; Smith, S.R.; Divoux, A.; Moresco, E.M.; et al. Insulin resistance and diabetes caused by genetic or diet-induced KBTBD2 deficiency in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E6418–E6426. [Google Scholar] [CrossRef] [PubMed]
- Saritas, T.; Cuevas, C.A.; Ferdaus, M.Z.; Kuppe, C.; Kramann, R.; Moeller, M.J.; Floege, J.; Singer, J.D.; McCormick, J.A. Disruption of CUL3-mediated ubiquitination causes proximal tubule injury and kidney fibrosis. Sci. Rep. 2019, 9, 4596. [Google Scholar] [CrossRef]
- Blondelle, J.; Shapiro, P.; Domenighetti, A.A.; Lange, S. Cullin E3 Ligase Activity Is Required for Myoblast Differentiation. J. Mol. Biol. 2017, 429, 1045–1066. [Google Scholar] [CrossRef]
- Papizan, J.B.; Vidal, A.H.; Bezprozvannaya, S.; Bassel-Duby, R.; Olson, E.N. Cullin-3-RING ubiquitin ligase activity is required for striated muscle function in mice. J. Biol. Chem. 2018, 293, 8802–8811. [Google Scholar] [CrossRef]
- Cirak, S.; von Deimling, F.; Sachdev, S.; Errington, W.J.; Herrmann, R.; Bönnemann, C.; Brockmann, K.; Hinderlich, S.; Lindner, T.H.; Steinbrecher, A.; et al. Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain 2010, 133, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, G.; Miyatake, S.; Lehtokari, V.L.; Todd, E.J.; Vornanen, P.; Yau, K.S.; Hayashi, Y.K.; Miyake, N.; Tsurusaki, Y.; Doi, H.; et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am. J. Hum. Genet. 2013, 93, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; O’Rourke, J.; Long, C.; Doering, J.; Ravenscroft, G.; Bezprozvannaya, S.; Nelson, B.R.; Beetz, N.; Li, L.; Chen, S.; et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Investig. 2014, 124, 3529–3539. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.A.; Ravenscroft, G.; Shaheen, R.; Todd, E.J.; Swanson, L.C.; Shiina, M.; Ogata, K.; Hsu, C.; Clarke, N.F.; Darras, B.T.; et al. Identification of KLHL41 Mutations Implicates BTB-Kelch-Mediated Ubiquitination as an Alternate Pathway to Myofibrillar Disruption in Nemaline Myopathy. Am. J. Hum. Genet. 2013, 93, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Martinez, A.; Cenik, B.K.; Bezprozvannaya, S.; Chen, B.; Bassel-Duby, R.; Liu, N.; Olson, E.N. KLHL41 stabilizes skeletal muscle sarcomeres by nonproteolytic ubiquitination. eLife 2017, 6, e26439. [Google Scholar] [CrossRef] [PubMed]
- Sambuughin, N.; Yau, K.S.; Olive, M.; Duff, R.M.; Bayarsaikhan, M.; Lu, S.; Gonzalez-Mera, L.; Sivadorai, P.; Nowak, K.J.; Ravenscroft, G.; et al. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am. J. Hum. Genet. 2010, 87, 842–847. [Google Scholar] [CrossRef]
- Papizan, J.B.; Garry, G.A.; Brezprozvannaya, S.; McAnally, J.R.; Bassel-Duby, R.; Liu, N.; Olson, E.N. Deficiency in Kelch protein KLHL31 causes congenital myopathy in mice. J. Clin. Investig. 2017, 127, 3730–3740. [Google Scholar] [CrossRef]
- Hannah, J.; Zhou, P. Distinct and overlapping functions of the cullin E3 ligase scaffolding proteins CUL4A and CUL4B. Gene 2015, 573, 33–45. [Google Scholar] [CrossRef]
- Sharma, P.; Nag, A. CUL4A ubiquitin ligase: A promising drug target for cancer and other human diseases. Open Biol. 2014, 4, 130217. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Raymond, F.L.; O’Meara, S.; Edkins, S.; Teague, J.; Butler, A.; Dicks, E.; Stevens, C.; Tofts, C.; Avis, T.; et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 2007, 80, 345–352. [Google Scholar] [CrossRef]
- Zhao, X.; Jiang, B.; Hu, H.; Mao, F.; Mi, J.; Li, Z.; Liu, Q.; Shao, C.; Gong, Y. Zebrafish CUL4a, but not CUL4b, modulates cardiac and forelimb development by upregulating TBX5a expression. Hum. Mol. Genet. 2015, 24, 853–864. [Google Scholar] [CrossRef]
- Zha, Z.; Han, X.R.; Smith, M.D.; Lei, Q.Y.; Guan, K.L.; Xiong, Y. Hypertension-associated C825T polymorphism impairs the function of Gbeta3 to target GRK2 ubiquitination. Cell Discov. 2016, 2, 16005. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yu, I.S.; Pai, C.H.; Lin, C.Y.; Lin, S.R.; Chen, Y.T.; Lin, S.W. Embryonic CUL4b is important for epiblast growth and location of primitive streak layer cells. PLoS ONE 2019, 14, e0219221. [Google Scholar] [CrossRef]
- Jiang, B.; Zhao, W.; Yuan, J.; Qian, Y.; Sun, W.; Zou, Y.; Guo, C.; Chen, B.; Shao, C.; Gong, Y. Lack of CUL4b, an E3 ubiquitin ligase component, leads to embryonic lethality and abnormal placental development. PLoS ONE 2012, 7, e37070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ye, J.; Chen, D.; Zhao, X.; Xiao, X.; Tai, S.; Yang, W.; Zhu, D. Differential expression profiling between the relative normal and dystrophic muscle tissues from the same LGMD patient. J. Transl. Med. 2006, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Chuang, K.W.; Yen, W.S.; Lin, S.Y.; Chen, H.H.; Chang, S.W.; Lin, Y.S.; Wu, W.L.; Tsao, Y.P.; Chen, W.P.; et al. Deficiency of nuclear receptor interaction protein leads to cardiomyopathy by disrupting sarcomere structure and mitochondrial respiration. J. Mol. Cell. Cardiol. 2019, 137, 9–24. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, Y. Cullin RING Ligase 5 (CRL-5): Neddylation Activation and Biological Functions. Adv. Exp. Med. Biol. 2020, 1217, 261–283. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiong, X.; Sun, Y. Cullin-RING Ligase 5: Functional characterization and its role in human cancers. Semin. Cancer Biol. 2020, 67, 61–79. [Google Scholar] [CrossRef]
- Min, K.D.; Asakura, M.; Shirai, M.; Yamazaki, S.; Ito, S.; Fu, H.Y.; Asanuma, H.; Asano, Y.; Minamino, T.; Takashima, S.; et al. ASB2 is a novel E3 ligase of SMAD9 required for cardiogenesis. Sci. Rep. 2021, 11, 23056. [Google Scholar] [CrossRef] [PubMed]
- Metais, A.; Lamsoul, I.; Melet, A.; Uttenweiler-Joseph, S.; Poincloux, R.; Stefanovic, S.; Valiere, A.; Gonzalez de Peredo, A.; Stella, A.; Burlet-Schiltz, O.; et al. ASB2alpha-Filamin a Axis Is Essential for Actin Cytoskeleton Remodeling during Heart Development. Circ. Res. 2018, 122, e34–e48. [Google Scholar] [CrossRef]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef]
- Diaz, S.; Wang, K.; Sjogren, B.; Liu, X. Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases. Biomolecules 2022, 12, 416. [Google Scholar] [CrossRef]
- Deeb, A.; Afandi, O.; Attia, S.; Fatih, A.E. 3-M syndrome: A novel CUL7 mutation associated with respiratory distress and a good response to GH therapy. Endocrinol. Diabetes Metab. Case Rep. 2015, 2015, 150012. [Google Scholar] [CrossRef]
- Pan, Z.Q. Cullin-RING E3 Ubiquitin Ligase 7 in Growth Control and Cancer. Adv. Exp. Med. Biol. 2020, 1217, 285–296. [Google Scholar] [CrossRef]
- Arai, T.; Kasper, J.S.; Skaar, J.R.; Ali, S.H.; Takahashi, C.; DeCaprio, J.A. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 9855–9860. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Kuwabara, H.; Arai, T.; Xiao, Y.; Decaprio, J.A. Disruption of the Fbxw8 gene results in pre- and postnatal growth retardation in mice. Mol. Cell Biol. 2008, 28, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Xie, B.; Martin, J.F. Targeting the Hippo pathway in heart repair. Cardiovasc. Res. 2022, 118, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.A.; Guo, S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J. Endocrinol. 2017, 233, R131–R143. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.; Hughes, J.; Hassan, A.B.; Bruning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef]
- Li, Z.; Pei, X.H.; Yan, J.; Yan, F.; Cappell, K.M.; Whitehurst, A.W.; Xiong, Y. CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity. Mol. Cell 2014, 54, 805–819. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Mani, D.C.; Satpathy, S.; Fereshetian, S.; Gasser, J.A.; Svinkina, T.; Olive, M.E.; Ebert, B.L.; Mertins, P.; Carr, S.A. Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 2020, 11, 359. [Google Scholar] [CrossRef]
- Sun, Z.; Xiao, W.D.; Li, N.K.; Chang, L.; Xu, P.; Li, Y.C. Large-Scale Profiling of Unexpected Tryptic Cleaved Sites at Ubiquitinated Lysines. J. Proteome Res. 2023, 22, 1245–1254. [Google Scholar] [CrossRef]
- Tan, B.; Peng, S.; Yatim, S.; Gunaratne, J.; Hunziker, W.; Ludwig, A. An Optimized Protocol for Proximity Biotinylation in Confluent Epithelial Cell Cultures Using the Peroxidase APEX2. STAR Protoc. 2020, 1, 100074. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Guo, J.; Wang, Z.; North, B.J.; Tao, K.; Dai, X.; Wei, W. Functional analysis of Cullin 3 E3 ligases in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fu, Y.; Peng, L.; Ogawa, Y.; Ding, X.; Rasband, A.; Zhou, X.; Shelly, M.; Rasband, M.N.; Zou, P. Immunoproximity biotinylation reveals the axon initial segment proteome. Nat. Commun. 2023, 14, 8201. [Google Scholar] [CrossRef]
- Keuss, M.J.; Thomas, Y.; McArthur, R.; Wood, N.T.; Knebel, A.; Kurz, T. Characterization of the mammalian family of DCN-type NEDD8 E3 ligases. J. Cell Sci. 2016, 129, 1441–1454. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Strategy | Cardiac Phenotype | Mechanism | Ref. |
|---|---|---|---|---|
| Nae1 | αMHCCre, mice | Ventricular non-compaction (E16.5), cardiac dysfunction (P1), and neonatal lethality (P3-7) | Accumulation of MST1 and LATS1/2; Hippo-YAP pathway inactivation | [34] |
| Csn8 | αMHCCre, mice | Necrosis and cardiac remodeling, heart failure, and premature lethality | Elevated p62 and LC3-II; impaired autophagosomes through regulation of Rab7 expression | [44] |
| Csn8 | αMHC-MerCre-Mer, mice | Necrosis and cardiac dysfunction | Impaired autophagosome removal and elevated oxidized protein levels | [45] |
| Rbx1 | Mouse gene trap model | Embryonic lethality (E7.5) | Proliferation defects due to p27 accumulation | [59] |
| Rbx1 | Tie2Cre, zebrafish | Dysregulated cardiac wall morphogenesis | Modulation of GLI1 levels in the endocardium | [60] |
| Rbx2 | Mouse gene trap model | Embryonic lethality (E11.5-12.5) | Defective vasculogenesis through accumulation of NF1 and Ras inhibition | [62] |
| Rbx2 | Tie2Cre, mouse | Embryonic lethality (E15.5) | Defective vasculogenesis and proliferation | [63] |
| Cul1 | Mouse gene trap model | Embryonic lethality (E6.5) during gastrulation | Accumulation of cyclin E | [66] |
| Cul3 | αMHCCre, mouse | Smaller body size, dilated right atrium and ventricle, severe cardiomyopathy, and atrial thrombi, followed by early neonatal lethality at P6 | Dysregulation of cardiac anti-oxidative and metabolic processes | [91] |
| Klhl40 | Mouse gene trap model | Severe nemaline myopathy | Loss of sarcomere thin filament proteins | [94] |
| Klhl41 | Antisense morpholinos, zebrafish | Neonatal lethality | Defective motor function and myofibrillar disorganization with nemaline body formation | [95] |
| Klhl41 | Mouse gene trap model | Nemaline myopathy | Disruption of sarcomeres and aberrant expression of muscle structural and contractile proteins resulting from prevention of nebulin aggregation and degradation | [96] |
| Klhl13 | CRISPR-Cas9 gene editing, mouse | Congenital myopathies | FLNC upregulation | [98] |
| Asb2 | Antisense morpholinos, zebrafish | Abnormal cardiac development | Regulation of Tbx2 expression through SMAD9 degradation | [110] |
| Asb2 | E2aCreVEC-Cre, mouse | Early death due to heartbeat defects | Regulation of FLNA degradation in immature cardiomyocytes at the onset of myofibrillogenesis | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zambrano-Carrasco, J.; Zou, J.; Wang, W.; Sun, X.; Li, J.; Su, H. Emerging Roles of Cullin-RING Ubiquitin Ligases in Cardiac Development. Cells 2024, 13, 235. https://doi.org/10.3390/cells13030235
Zambrano-Carrasco J, Zou J, Wang W, Sun X, Li J, Su H. Emerging Roles of Cullin-RING Ubiquitin Ligases in Cardiac Development. Cells. 2024; 13(3):235. https://doi.org/10.3390/cells13030235
Chicago/Turabian StyleZambrano-Carrasco, Josue, Jianqiu Zou, Wenjuan Wang, Xinghui Sun, Jie Li, and Huabo Su. 2024. "Emerging Roles of Cullin-RING Ubiquitin Ligases in Cardiac Development" Cells 13, no. 3: 235. https://doi.org/10.3390/cells13030235
APA StyleZambrano-Carrasco, J., Zou, J., Wang, W., Sun, X., Li, J., & Su, H. (2024). Emerging Roles of Cullin-RING Ubiquitin Ligases in Cardiac Development. Cells, 13(3), 235. https://doi.org/10.3390/cells13030235