1. Introduction
The immune system protects an organism from diseases of various pathogens, e.g., viruses, bacteria, and fungi [
1]. This protection depends on the ability to discriminate between the host (self) and external stimuli (non-self), as well as on directing a response to external stimuli to protect the body from harmful pathogens [
2,
3]. Innate immunity provides a rapid, non-specific defense by recognizing general molecular patterns on pathogens through pattern recognition receptors, which detect common structures such as bacterial cell wall components, triggering an immediate immune response. Adaptive immunity, on the other hand, offers a more targeted and specific defense by recognizing unique antigens via T and B cells, which develop memory for future encounters. Together, innate and adaptive immunity ensure effective detection and elimination of foreign invaders. Empowering the immune system to clear conditions which could not be resolved, such as cancer or autoimmune diseases, has gained attention. Coming from this idea, cellular immunotherapy and, in particular, chimeric antigen receptor (CAR)-expressing T cell therapy has provided meaningful clinical responses in many patients by redirecting T cells to target cancer cells and other pathogenic cells, such as HIV-specific CAR T cells [
4,
5,
6].
CAR T cells are genetically modified to express an artificial receptor which combines different aspects of the physiological T cell receptor functions with a defined extracellular antigen binding domain. The CAR is modularly designed with at least one element mediating specificity towards the target antigen. Commonly, this is a single-chain variable fragment (scFv) derived from an antibody. A hinge connects the binding domain with the transmembrane part, as well as the intracellular components of the CAR, i.e., an activation domain, which is often CD3zeta, and at least one co-stimulatory domain, e.g., based on 4-1BB or CD28 [
7,
8].
When expressed by T cells, functional CAR constructs endow these immune cells with specific effector functions upon antigen encounter. Several seminal hallmarks of functional CAR T cells have been proposed [
9,
10]. After, specific CAR-dependent target recognition cytokines are produced, e.g., IFN-γ and IL-2, supporting cytotoxicity as well as CAR T cell expansion. However, the clinical cancer setting is considered more complex. It was proposed that reinvigorated T cell effector function, upon repeated antigen exposure, can serve as an in vitro surrogate to mimic the challenging patient situation [
11,
12,
13].
Besides the promises and the impressive clinical data of CAR T cell therapy, limitations exist, which need to be overcome to further improve cellular immunotherapy for patients [
14,
15,
16]. For instance, anti-tumor function, as well as persistence of CAR T cells, can be impacted by an anti-CAR immune reaction by the host [
17,
18]. Cellular and/or humoral immunogenicity of the CAR may be related to non-human protein sequences included in the CAR. Especially the use of scFv sequences, which are often derived from murine monoclonal antibodies, may pose the risk of a human anti-mouse immune reaction [
19]. Recent reports describe the presence of anti-CAR antibodies and anti-CAR TCRs in some patients treated with CAR T cells, particularly when CAR T cells were applied multiple times during treatment or when different CAR products were administered in sequential treatment lines [
20,
21].
Another limitation of CAR T cell therapy is related to the target antigen expressed on non-malignant cells. Although extensive preclinical characterization of CAR T cells is performed, unexpected toxicities were reported for several CAR constructs [
22]. For example, anti-carbonic anhydrase IX (CAIX) CAR T cells caused liver toxicities in patients with metastatic renal cell carcinoma [
23,
24]. Patients with carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5)-positive solid tumors suffered from serious adverse events such as gastrointestinal, but also pulmonary, toxicity after infusion of anti-CEACAM5 CAR T cell treatment [
25]. Patients receiving anti-claudin 18.2 (CLDN18.2) CAR T cells developed mucosal toxicity [
26]. Epidermal growth factor receptor (EGFR)-directed CAR T cells were reported to cause mucosal and cutaneous adverse events [
27,
28]. Finally, patients treated with receptor tyrosine-protein kinase erbB-2 (ERBB2 or HER2)-targeting CAR T cells experienced cardiopulmonary toxicity [
29].
To mitigate the risk of such toxicities, tissue cross-reactivity studies are usually performed [
30,
31,
32]. These screening assays routinely utilize immunohistochemistry to identify non-specific and specific binding of antibodies or antibody-like proteins, in different types of human or animal tissues, respectively. Unspecific binding occurs between molecules that are not intended to interact, which can mislead decision making. One approach is the use of negative controls to establish the baseline binding level in the absence of the specific target. For instance, in antibody-based assays, including a control without the specific antigen helps identify whether the antibody binds non-specifically to other components in the sample. In immunofluorescence, elevated background fluorescence can be indicative for unspecific binding, which can be assessed using isotype controls. Additionally, in IHC, employing secondary antibody only controls can help confirm the specificity of binding. Noteworthy, the staining profiles, which are indicative for protein expression, of animal and human tissues may differ in intensity and distribution, e.g., angiotensin-converting enzyme 2, ACE2, the receptor for the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is expressed in human cardiomyocytes and not in murine cardiomyocytes [
33]. Tissue cross-reactivity studies support the preclinical safety assessment data package.
Although conventional immunohistochemistry is widely used for tissue cross-reactivity studies, this method has some constraints, including high inter-observer variability and the use of only one single marker per tissue section [
34]. In contrast, immunofluorescence (IF) offers higher sensitivity and allows for multicolor labeling, enabling the simultaneous detection of multiple proteins within a single sample. Additionally, IF provides better spatial resolution and less background staining, making it easier to visualize and quantify specific proteins in complex tissues or cells. Particularly, multiplexing and semi-automated analysis approaches can help to overcome the limitation of testing a single marker per tissue section. This improvement is of particular interest for the analysis of novel antibody fragments since the analysis of the binders and additional reference antibodies to characterize the cellular composition of the tissue can be performed on the same tissue slice. Additionally, multiplexing enables data generation from samples with limited size or scarce availability.
As an additional approach, to identify off-tumor and/or off-target toxicities in vivo, experiments in mice are conducted. However, many CAR T cells are designed with antigen-binding domains selectively recognizing human epitopes, but not murine antigens, therefore, providing only limited information on potential off-tumor targeting of these CAR T cells.
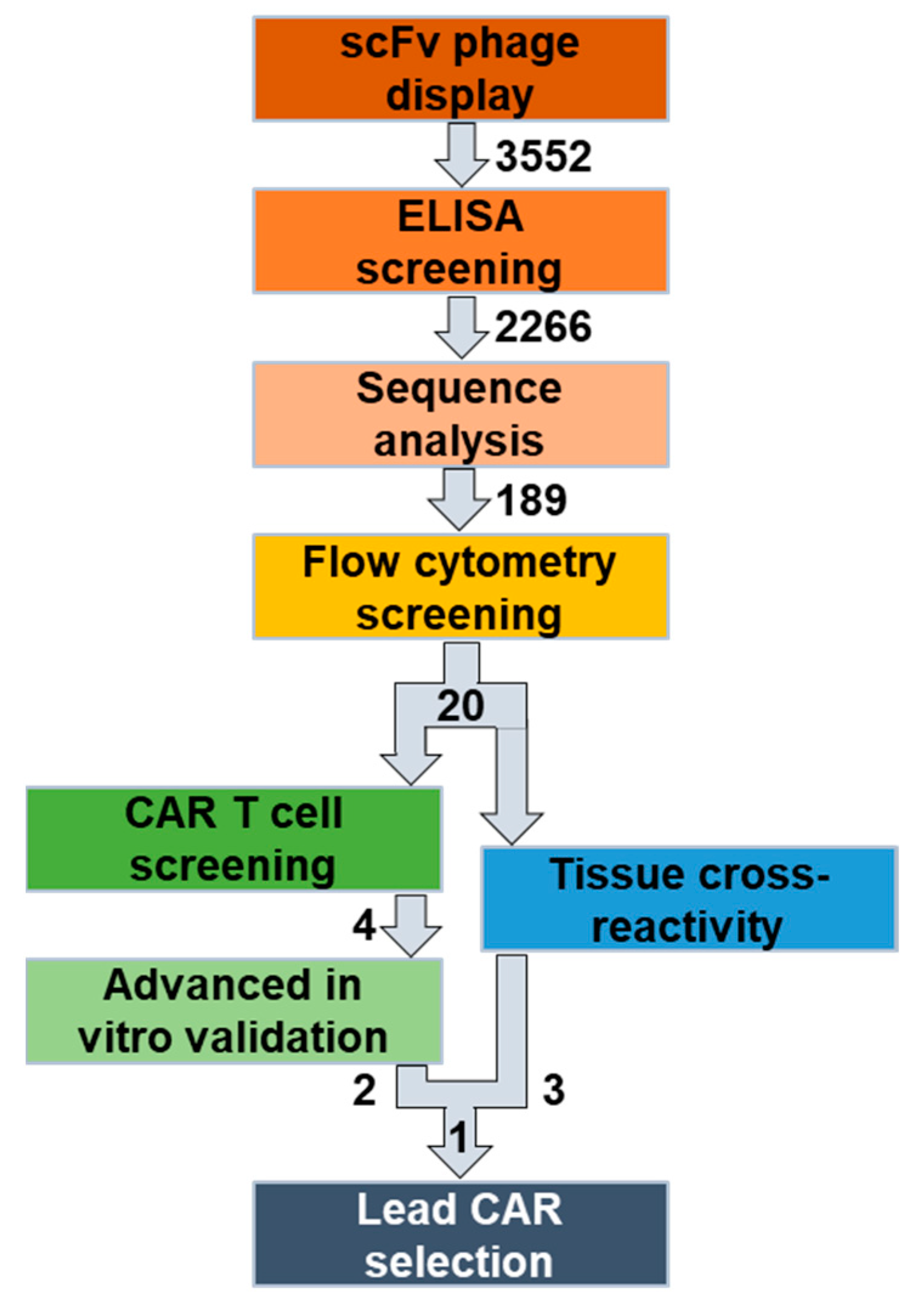
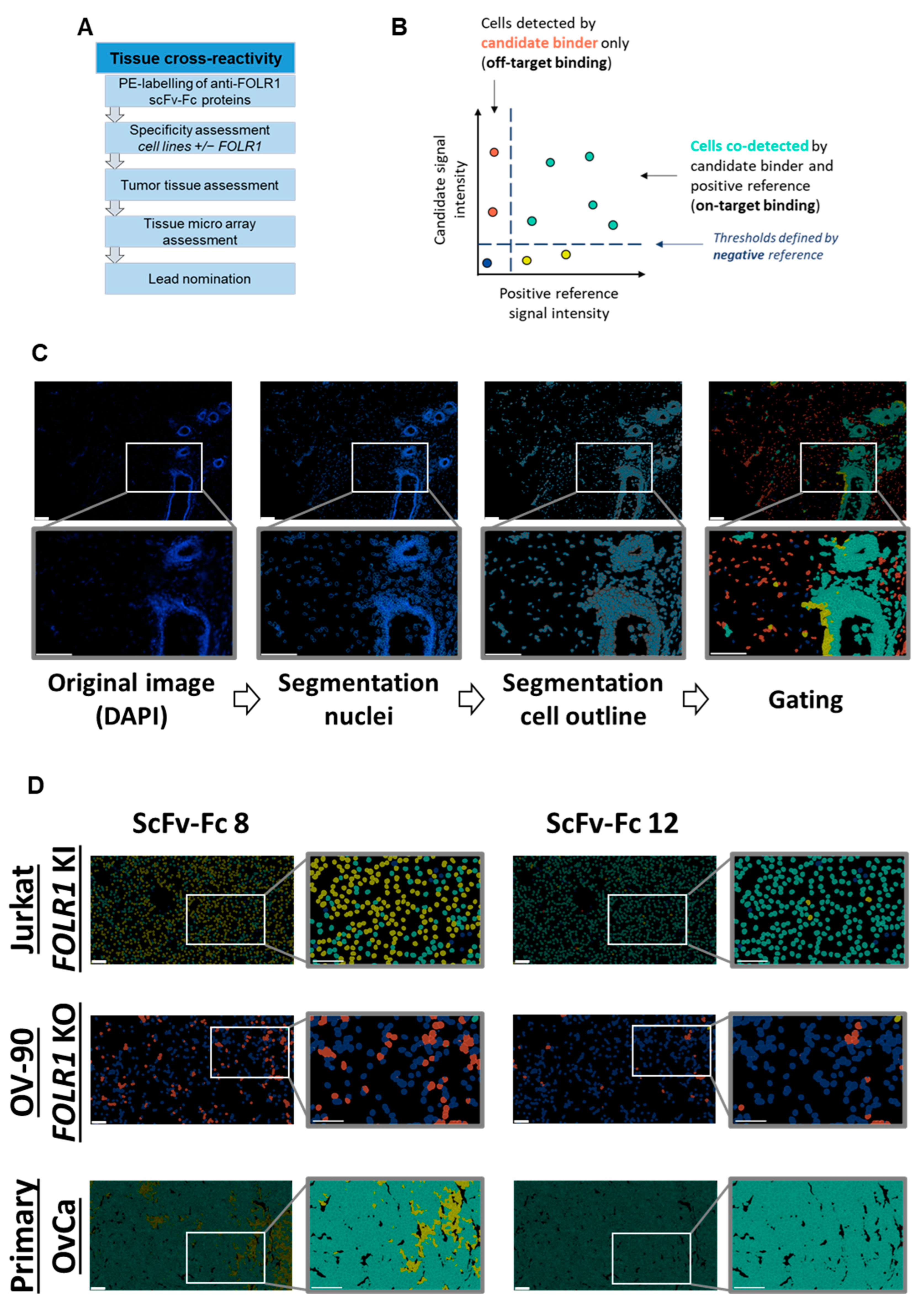
To overcome these limitations, we established a novel workflow to identify and characterize novel, fully human CAR candidates (
Figure 1). Here, we selected folate receptor 1 (FOLR1 or FRα) as the target antigen for the treatment of ovarian cancer [
35,
36]. We screened a naïve, human B cell receptor library to generate fully human scFv sequences specific for human and murine FOLR1, to be able to anticipate potential off-tumor reactivity in mouse studies. Since the folate receptor family has a high degree of similarity [
37], we assessed specificity of the new human/murine FOLR1-directed binders towards other human/murine folate receptor variants hFOLR2, hFOLR3, hFOLR4, mFOLR2, and mFOLR4, respectively.
During the initial screening and panning campaigns, we performed positive and negative selections to isolate FOLR1-specific scFvs. Subsequently, FOLR1-binding domains were assessed as scFv-Fc fusion proteins in flow cytometry for advanced specificity characterization against members of the human and murine FOLR families. Next, FOLR1-specific binder sequences were transferred into second generation CAR constructs, and a functional in vitro high-throughput screening was performed, employing the FOLR1-proficient ovarian cancer cell line OV-90 as well as a CRISPR/Cas9-mediated FOLR1-deficient ovarian cancer cell line OV-90
FOLR1 KO [
36]. The high-throughput screening assay is based on a co-culture of CAR T cells with target cells and repetitive addition of fresh target cells. Automated plating of target cells and CAR T cells enabled the parallel analysis of all the CAR constructs simultaneously. The best functional performance of CAR candidates in vitro was determined by target cell lysis, cytokine release, and CAR T cell expansion employing automation approaches. Subsequently, FOLR1-targeting CAR candidates were further characterized in in vitro co-culture experiments under more challenging conditions, i.e., unfavorable effector to target cell ratios as well as repetitive target cell exposure. Finally, different ovarian cancer cell lines were employed as target cells in in vitro co-culture experiments, namely OV-90, OV-90
FOLR1 KO, OVCAR-3, and SKOV-3.
In order to complement the anti-FOLR1 scFv characterization, we set up tissue cross-reactivity experiments employing a multiplexing imaging system. We used anti-FOLR1 scFv-Fc fusion proteins in an automated, high-plex imaging approach to analyze the respective staining profiles relative to a well-characterized anti-FOLR1 monoclonal antibody, LK26 [
38,
39], and to assess on-tumor and off-tumor binding on healthy human tissues (adrenal gland, ovary, pancreas, thyroid, cerebellum, cerebrum, lung, spleen, uterus, cervix, breast, placenta, heart, skin, skeletal muscle, kidney, stomach, small intestine, liver, and salivary gland) and malignant human tissues (high grade serous ovarian cancer).
Taken together, we established a new workflow to identify and characterize novel, fully human and functional CAR candidates against FOLR1, based on functional as well as tissue reactivity data.
2. Materials and Methods
2.1. Identification of Antibody Fragments
Two human, naïve scFv phage display libraries, constructed at Miltenyi Biotec, were used for the identification of novel binders against FOLR1. These libraries differed in the variable domains of the light chain, as they contained either the κ or the λ isotype. The library was constructed from naïve human B cells of more than 120 healthy donors, resulting in an antibody diversity of 5 × 1010. For identification of antibody fragments, mouse FOLR1 (mFOLR1), human FOLR1-hIgG1Fc (hFOLR1-Fc), and human FOLR2 (hFOLR2) were purchased from Acro Biosystems. Human FOLR3 was purchased from R&D systems, and human IgG1-Fc (hIgG1Fc) was purchased from amsbio. For panning, antigens were directly immobilized on 96-well MaxiSorb ELISA Plates (Nunc) for 1 h at RT, followed by blocking with 1% (w/v) BSA in PBST (PBS supplemented with 0.05% Tween 20) for 1 h at RT. For each strategy, the κ and λ library was preincubated in BSA blocked wells to remove unspecific binding antibody phage particles, before incubation for 2 h on immobilized antigens of interest. Competing antigens were added directly after transferring the library to the antigen of interest. After stringent ELISA washing, scFv phage particles were eluted by adding 10 µg/mL trypsin and incubating for 30 min at 37 °C. Eluted phage particles were used to infect E. coli TG1 (O.D.600 0.4–0.5) and phage production was facilitated using M13K07 helper phage. After the third panning round, eluted phage particles were used to infect E. coli TG1 (O.D.600 0.4–0.5). Resulting single cell colonies were used to inoculate 96 well PP microtiter plates (Greiner BioOne, Kremsmuenster, Austria). The next day, 10 µL cultures were used to inoculate plates for expression of soluble scFv antibody fragments.
Expressed scFvs were analyzed for their capability to bind to hFOLR1-Fc, mFOLR1, hFOLR2, or hIgG1Fc in ELISA. A total of 30 ng antigen per well was coated using 384 well high binding PS microtiter plates (Greiner BioOne, Kremsmuenster, Austria), in PBS over night at 4 °C, and subsequent blocking was performed with 2% BSA-PBST (2% (w/v) BSA in PBST) for 1 h at RT. After three washing steps using an ELISA washer (EL406, BioTek), antibodies containing overnight cultures mixed with anti c-myc-HRP detection antibodies (Miltenyi Biotec, Bergisch Gladbach, Germany) were incubated for 1 h at RT. Finally, 30 µL of TMB (3,3′,5,5′-tetramethylbenzidine) was added and the color reaction was stopped by adding 30 µL 1 N sulfuric acid. Absorbances were measured at 450 nm (with 630 nm reference wavelength) using an ELISA reader (EPOCH, BioTek, Winooski, VT, USA).
2.2. Cloning of scFv-Fc Fusion Proteins
ScFv genes were cloned as scFv-Fc fusion into mammalian expression vector, based on pFUSE, InvivoGen (San Diego, CA, USA). Therefore, the scFv gene was amplified via PCR out of the phage display vector. ScFv genes and backbone vector were digested with NcoI/NotI and the backbone vector was additionally digested with Shrimp Alkaline Phosphatase (New England BioLabs, Ipswich, MA, USA) to avoid religation. NEB® Stable Competent E. coli (High Efficiency) was used for transformation of the ligation mix. ScFv sequences were validated by Sanger sequencing.
2.3. Expression and Purification of Anti-FOLR1 scFv-Fc Candidates
Selected binders were transiently expressed as antibody fragments in the scFv-Fc format and subsequently purified via Protein A. Purity was confirmed via SDS page and protein concentrations were determined using a microplate reader by measuring UV absorbance at 280 nm. Purified proteins were stored at −20 °C until further processing.
2.4. Tissue Processing for Microscopy
We recently detailed our tissue processing method [
40]. Briefly, frozen embedded tissue samples were sectioned at 8 µm thickness using a Leica CM3050 cryostat, and mounted on Menzel SuperFrost Plus slides (Thermo Fisher Scientific, Waltham, MA, USA). For acetone fixation, sections were fixed in acetone at −20 °C for 3 min, then stored at −80 °C. On the day of use, slides were immersed in acetone at −20 °C for 10 min to thaw, then briefly air-dried. The appropriate MACSwell™ Imaging Frame was immediately mounted on each slide, and the specified initial volume of MACSima™ Running Buffer was added, as outlined in the MACSwell™ Imaging Frame data sheet. For PFA fixation, cryo-sectioned slices were stored directly at −80 °C. On the day of use, slides were incubated in a 4% PFA solution at room temperature for 10 min and washed three times with MACSima™ Running Buffer. After washing, the MACSwell™ Imaging Frame and initial MACSima™ Running Buffer volume were added per the data sheet instructions. A preliminary DAPI staining was then performed: the sample was stained for 10 min with a 1:10 dilution of DAPI staining solution (volume according to the working format of the MACSwell™ Imaging Frames). Following this, the DAPI solution was removed, and three washes with MACSima™ Running Buffer were conducted. Finally, the initial volume of MACSima™ Running Buffer was added to the sample.
2.5. Cyclic Immunofluorescence Staining with the MACSima Imaging Platform
We have previously reported the use of the MACSima™ Imaging Platform [
40]. The MACSima™ Imaging System is a fully automated instrument that integrates liquid handling with widefield microscopy for cyclic immunofluorescence imaging. Briefly, each staining cycle involves automated steps of immunofluorescent staining, sample washing, multifield imaging, and signal removal (either through photobleaching or REAlease Reagents). Cyclic immunofluorescence with the MACSima™ Imaging System is ideally performed on thin cryosections (a few microns thick), cultured cells, or suspension cells (either captured in microcavities or centrifuged onto glass). To enable MACSima™ imaging cyclic staining (MICS) experiments, we developed MACSwell™ Sample Carriers, which provide reaction cavities for the MACSima™ Imaging Platform. We also designed various sample carriers—MACSwell™ One, Two, and Four Imaging Frames, as well as MACSwell™ 24 Imaging Plates—to accommodate tissue sections of different sizes or adherent cells.
2.6. PE Labeling of scFv-Fc
ScFv-Fc proteins were coupled to Succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (SMCC)-activated PE (Agilent, Cat-No: PB31). Labeling was performed on the semi-automated cell separator MultiMACS™ (Miltenyi Biotec, Bergisch Gladbach, Germany) using MACS® Protein A and G MicroBeads and MACS® Columns (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer.
2.7. MACSima™ Data Analysis
Tumor marker FOLR1 expression was quantified on a single-cell level on primary ovarian cancer tissue. Image data sets were segmented using MACS® iQ View Software version 1.3.0 (Miltenyi Biotec, Bergisch Gladbach, Germany) based on nuclei, i.e., DAPI signal, and epithelial cell membrane marker EpCAM, identifying individual cells. Single cells were analyzed for FOLR1 expression. To identify single cells, the DAPI channel was used to segment cell nuclei and a distinct area around the nuclei as the cytoplasm. All features (cell size, nucleus size, biomarker expression, etc.) were measured in all channels. The mean intensities of all channels were calculated from the extracted data. Using MACS® iQ Views plot tab feature, scatter plots of the positive reference versus the candidate binders were plotted, and the mean intensity of the negative binder was used as a cut-off value to set gates on the mentioned plots. The aim was to detect specific signals on single cells and, in particular, cells that were co-stained with the positive reference and the candidate binder, i.e., the double positive cells.
2.8. Generation of CAR Plasmids
Plasmids encoding the CAR constructs were prepared using standard molecular biology and cloning techniques. Human naïve single-chain variable fragments (scFv), identified after panning, were cloned into a lentiviral vector (Miltenyi Biotec, Bergisch Gladbach, Germany) using AscI and EcoRV. Construct configuration was scFv (Vh-Vl orientation)—human CD8alpha hinge—CD8alpha transmembrane domain—human 4-1BB costimulatory domain—human CD3 zeta domain. The scFv contained a (G4S)3 linker domain. Lentiviral vectors contained a truncated human low-affinity nerve growth factor receptor (ΔLNGFR) expression cassette separated by a P2A element for co-expression as a transduction marker.
2.9. Generation of CAR T Cells
The generation of CAR T cells has been previously described [
41]. In summary, peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples via density gradient centrifugation. T cells were purified from PBMCs using the Pan T Cell Isolation Kit, human (Miltenyi Biotec, Bergisch Gladbach, Germany), and activated in TexMACS™ Medium (Miltenyi Biotec, Bergisch Gladbach, Germany) supplemented with T Cell TransAct™ (Miltenyi Biotec), along with 12.5 ng/mL of recombinant human interleukin IL-7 and 12.5 ng/mL of recombinant human IL-15 (both from Miltenyi Biotec, Bergisch Gladbach, Germany). Twenty-four hours after activation, T cells were transduced at a multiplicity of infection (MOI) of 5 using vesicular stomatitis virus glycoprotein G (VSV-G) pseudotyped lentiviral supernatants. Lentiviral particles were produced in-house by providing the required viral genes (gag/pol, rev and VSV-G) in trans, which was performed by co-transfection of 293T producer cells with four plasmids. Three days post-activation, T Cell TransAct™ was removed from the medium, and the cells were cultured in TexMACS™ Medium containing 12.5 ng/mL each of recombinant human IL-7 and IL-15. After an additional 9 to 12 days, CAR T cells were used for in vitro assays. On the day of assay use, the number of viable CAR T cells was determined by staining them with 7-AAD, biotinylated human FOLR1 Protein (Miltenyi Biotec, Bergisch Gladbach, Germany), and an anti-biotin antibody conjugated to PE.
2.10. Cell Lines and Culture Conditions
Human embryonic kidney 293T cells (HEK293T, ACC 635) were obtained from the DSMZ—German Collection of Microorganisms and Cell Cultures. OV-90 (CRL-11732), NIH:OVCAR-3 (HTB-161), and SKOV-3 (HTB-77) cells were obtained from ATCC and cultured as recommended. Jurkat E6 cells were cultured at 37 °C, 5% CO2 in RPMI with 10% FCS and 5 mM L-glutamine. Cell lines were routinely analyzed for mycoplasma contamination.
2.11. Cell Line Generation
OV-90 FOLR1 KO cells were created using CRISPR/Cas9 followed by single-cell cloning. Ribonucleoprotein complexes were prepared according to the manufacturer’s instructions (IDT, Coralville, IA, USA). Briefly, CRISPR-Cas9 gRNA targeting FOLR1 (sequence: CCTACCTATATAGATTCAACTGG) was combined with CRISPR-Cas9 crRNA in a 1:1 ratio, incubated for 5 min at 95 °C, then, allowed to cool at RT for 20 min to form the gRNA complex. Next, the gRNA complex was incubated with Cas9 nuclease at a 1:1 ratio for 20 min at RT to form ribonucleoprotein complexes. Subsequently, 1 × 106 OV-90 cells were resuspended in 40 µL of electroporation buffer (Miltenyi Biotec, Bergisch Gladbach, Germany), including 10 µL of ribonucleoprotein complexes (2.7 µg total), and transferred to an electroporation cuvette (Miltenyi Biotec, Bergisch Gladbach, Germany). Electroporation was performed using the CliniMACS® Electroporator (Miltenyi Biotec). Afterward, cells were cultured under standard conditions, and FOLR1 KO cells were sorted with the MACSQuant® Tyto® Cell Sorter (Miltenyi Biotec, Bergisch Gladbach, Germany). Knock-out efficiency was assessed through flow cytometry, qPCR, microscopy, and gDNA sequencing to confirm FOLR1 deletion. Single-cell clones were obtained by serial dilution.
Jurkat cells expressing various FOLR variants were generated via lentiviral transduction. Lentiviral particles encoded human FOLR1 (gene ID: 2348), human FOLR2 (gene ID: 2350), human FOLR3 (gene ID: 2352), human FOLR4 (gene ID: 390243), murine FOLR1 (gene ID: 14275), murine FOLR2 (gene ID: 14276), or murine FOLR4 (gene ID: 64931), respectively.
2.12. Cytotoxicity Assays
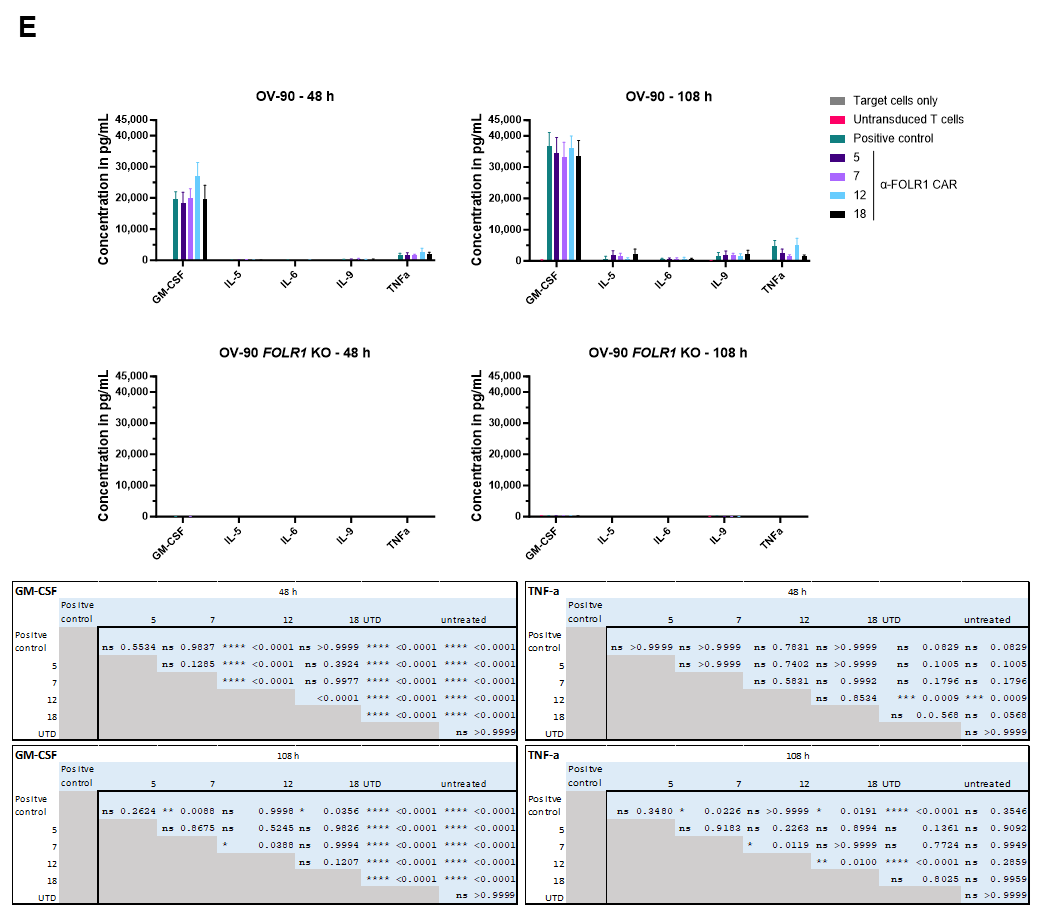
Target cells were seeded in triplicate in 96-well plates at densities of 2 × 104 ovarian cancer cells, or 1.5 × 104 Jurkat cells per well, in their respective culture media. CAR T cells were added 18 h later at an effector-to-target (E:T) cell ratio of 2:1, with the final well volume adjusted to 200 μL. The total T cell count was matched to that of the CAR T cell group with the lowest transduction efficiency. Cytotoxicity was assessed by monitoring the reduction in green surface area using the IncuCyte® S3 Live-Cell Analysis System (Sartorius, Gottingen, Germany) and the associated IncuCyte® software (versions v2017A, v2018C, and v2019A). The decrease in green surface area was calculated relative to baseline. At the end of the 48 h co-culture period, CAR T cells were analyzed by flow cytometry for activation markers (CD25, CD69, 4-1BB), exhaustion markers (TIM3, LAG3, PD1), and phenotype markers (CD62L, CD45RO, CD45RA). Additionally, after 24 h, 50 μL of medium was collected for cytokine analysis using the MACSPlex Cytokine 12 Kit, human (Miltenyi Biotec, Bergisch Gladbach, Germany). Then, 2 × 104 GFP-expressing cells were seeded and co-cultured with 1 × 104 anti-FOLR1 CAR T cells to measure antigen-dependent lysis of ovarian cancer cells for five days. After 48 h of co-culture, 2 × 104 ovarian cancer cells were added to the co-culture. Cell lysis by the CAR T cell candidates was tracked by measurement of the green area confluency. As negative controls, ovarian cancer cells were cultured without addition of CAR T cells or with untransduced T cells. A positive control CAR T cell construct, known to successfully induce FOLR1-specific lysis of target cells, was included. The top four candidates identified by the CAR T cell screening were included in this assay, comprising three donors each. After 48 h and 108 h, flow cytometric measurement of activation marker expression 4-1BB and CD69 was performed. In addition, activation markers on a fourth donor were measured upon co-culture with FOLR1low (OVCAR-3)-expressing cells.
2.13. Repetitive Killing Assays
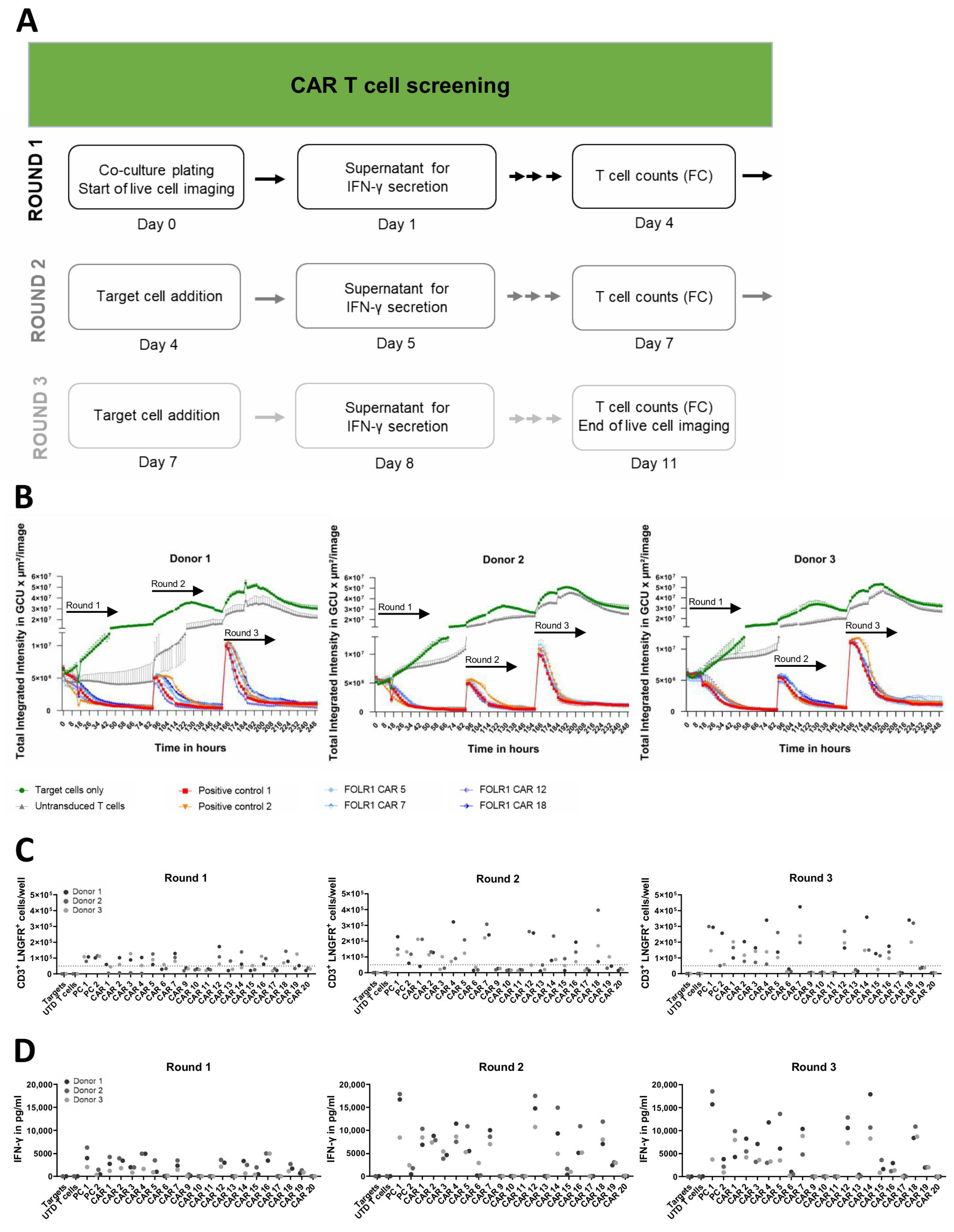
Lentiviral particles were used to transduce primary human T cells of three independent donors for CAR expression with an MOI of 5. Frequency and absolute numbers of CAR T cells were determined by quantification of CD3+ LNGFR+ events using flow cytometry. CAR T cells were cultured at an E:T ratio of 2.5:1 with 2 × 104 OV-90 or OV-90 FOLR1 KO cells, which were transduced to express GFP. As controls, CAR T cells were cultured in absence of target cells, and OV-90 or OV-90 FOLR1 KO cells were cultured in absence of CAR T cells. Additionally, OV-90 and OV-90 FOLR1 KO cells were cultured with untransduced T cells or with T cells expressing one of two positive control CAR constructs, respectively. After 24 h of cell culture, supernatants were analyzed for cytokine secretion, e.g., IFN-γ, by using the MACSPlex Cytokine 12 Kit, human (Miltenyi Biotec, Bergisch Gladbach, Germany). After 92 h, CAR T cells (CD3+ LNGFR+) were quantified by flow cytometry. In addition, 2 × 104 fresh OV-90 and OV-90 FOLR1 KO cells were added, according to the experimental setup, to initiate the second round of co-culture. Twenty-four hours later, supernatants were again analyzed for IFN-γ secretion. After 164 h, 5 × 104 CAR T cells (CD3+ LNGFR+) were quantified by flow cytometry. The third round of co-culture was started by addition of fresh OV-90 and OV-90 FOLR1 KO cells, according to the experimental setup. Twenty-four hours later, supernatants were again analyzed for IFN-γ secretion. After 260 h, CAR T cells (CD3+ LNGFR+) were quantified by flow cytometry. The total duration of three rounds of co-culture was 11 days, and cytotoxicity towards OV-90 and OV-90 FOLR1 KO cells, respectively, was quantified over time every 2 h by acquisition of the GFP signal over time by measuring the Total Integrated Intensity as Green Calibrated Units per µm2/image (IncuCyte S3, Sartorius, Gottingen, Germany).
2.14. Flow Cytometry
Target specificity of selected anti-FOLR1 scFv-Fc candidates was validated in flow cytometry using cell lines that were transduced with the respective FOLR variant. For multiplexing, non-target cells were labeled with CellTrace™ Violet or Cell Trace™ FarRed (ThermoFisher, Waltham, MA, USA) according to manufacturer’s protocol. Next, 1 × 105 cells per cell line were seeded into a 96-well plate and cells were stained with 1 µg scFv-Fc for 30 min at 4 °C. Upon two washing steps, secondary antibody staining was performed using AffinitPure F(ab’)2 Fragment Goat Anti-Human IgG-PE (Jackson ImmunoResearch, Cambridgeshire, UK) for 10 min at 4 °C.
Antibody conjugates that were used for surface marker expression are listed in
Supplementary Table S3. Stainings performed with antibody conjugates from Miltenyi Biotec (Bergisch Gladbach, Germany) were performed as recommended by the supplier. Antibodies from Biolegend were applied at a concentration of 5 µg/mL and incubated for 10 min at 4 °C, followed by a successive washing step. To exclude dead cells, 7-AAD or PI was used. Samples were acquired at the MACSQuant
® Analyzer 10 or 16. MACSQuantify™ Software v2.13.0/v2.11.0, and Flow Logic software 7.2.1 were used for data analysis.
For quantification of CAR T cell expansion in repetitive killing assays, samples were taken at the end of each round of co-culture. After incubation with antibodies in PEB (CliniMACS® PBS/EDTA buffer supplemented with 0.5% MACS® BSA stock solution), the cell suspension was diluted by the addition of PEB plus Poloxamer 188 solution. These samples were acquired on a MACSQuant® X Flow Cytometer (Miltenyi Biotec, Bergisch Gladbach, Germany).
2.15. Statistical Analysis
Unless indicated otherwise, all graphs display the mean, with error bars representing the standard error of the mean. Statistical analyses were performed with GraphPad Prism 8.
2.16. Primary Human Blood and Tissues
High-grade serous ovarian cancer patient samples were purchased from BioIVT or provided by Prof. Dr. Peter Mallmann (Department of Obstetrics and Gynaecology, Faculty of Medicine, University of Cologne), with all samples obtained under informed patient consent. FDA Standard Frozen Tissue Array—Human Adult Normal was purchased from Biocat (Heidelberg, Germany), including adrenal gland, ovary, pancreas, thyroid, cerebellum, cerebrum, lung, spleen, uterus, cervix, breast, placenta, heart, skin, skeletal muscle, kidney, stomach, small intestine, liver, and salivary gland.
2.17. Ethics Approval
For all studies involving human primary ovarian cancer tissue, written informed consent was obtained in accordance with the University Hospital Cologne Review Board’s approved protocol. Healthy whole blood samples were collected from donors who had provided prior written consent. Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats obtained from anonymous healthy donors, purchased from the German Red Cross in Dortmund. All blood samples were handled following required ethical and safety guidelines.
4. Discussion
Cellular immunotherapy has provided substantial clinical benefits to many patients. Particularly, the use of CAR T cells for the treatment of hematological cancers, but also autoimmune diseases, has improved patient survival significantly [
49,
50,
51]. However, several aspects in the generation and characterization of a CAR can be optimized to further improve the clinical benefit.
We initially hypothesized that several limitations of current CAR T cell approaches, e.g., immunogenicity and off-tumor reactivity, may be addressed early in the development process by comprehensive and complementary assays. As a proof of concept in this study, we set up a new workflow for the identification and selection of CAR candidates based on functionality and on-target/off-tumor reactivity.
Here, we report, (i) the generation of highly specific, fully human scFv sequences from a naïve human B cell library against FOLR1, (ii) the functional high-throughput in vitro assessment of anti-FOLR1 CAR T cells based on these fully human scFv sequences, and (iii) the tissue cross-reactivity characterization of the anti-FOLR1 scFvs to assess off-target and on-target/off-tumor binding.
A fully human CAR design, including extra- and intracellular domains, may decrease the risk of immunogenicity and may consequently improve CAR T cell therapies. The origin and composition of the CAR sequence used, e.g., murine sequences, may induce an immune response in patients after CAR T cell infusion. This anti-CAR immune response may hamper efficacy as well as persistence of CAR T cell treatment [
17,
18]. Therefore, sequences of murine origin are frequently humanized to mitigate the risk of immunogenicity. Alternatively, fully human sequences from human-derived sources almost completely remove the risk of cellular and/or humoral immunogenicity [
21,
52,
53]. In addition to individual studies reporting human anti-mouse immune reactions, the potential issue of immunogenicity may be even more relevant when CAR T cells are injected multiple times or when different CAR products are applied to a patient.
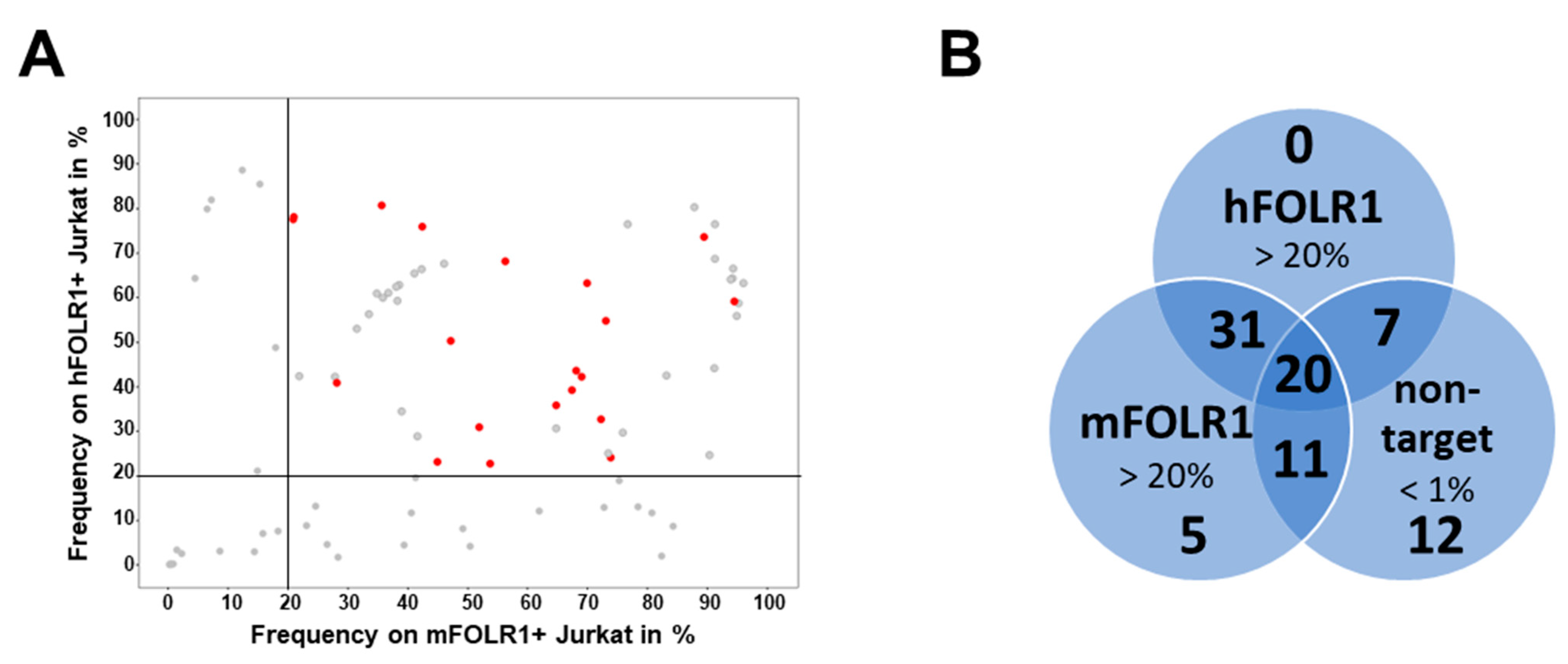
Consequently, to retrieve fully human scFv sequences, we employed a newly designed human naïve scFv library from B cells. In addition, panning strategies were designed to select highly specific CAR candidates. To isolate antibody fragments recognizing both mouse and human FOLR1, different panning strategies, using either one or both of the targets in consecutive selection rounds, were evaluated. As expected, most antibody hits were identified when both antigens were combined, but also a decent number of cross-reactive clones was identified from the panning strategy, with only mFOLR1. Potentially, dimerization of the target protein and presence of the Fc domain in the human target protein influenced the selection of binders. In summary, alternating murine and human target protein in a phage display selection led to the identification of a high number of cross-reactive antibody fragments.
Additionally, we performed extensive flow cytometric assays to characterize scFv specificity against human and murine FOLR variants. This step addressed scFv binding to its cognate antigen in a physiological context, i.e., full length protein with potential post-translational modifications, without sample fixation prior to assessment. To analyze proper target specificity in a cellular context, we evaluated target recognition on transduced cell lines expressing human and murine FOLR variants. Previous work has shown that CAR functionality is impacted by receptor affinity, whereby both positive and negative effects of affinity tuning have been seen for CAR T cell functionality [
54,
55]. Therefore, binder selection was not narrowly restricted to high staining frequencies on target cells, but a broad range of scFv-Fc candidates was selected for further evaluation in the CAR format. To select highly specific CAR candidates, they were not only positively selected for hFOLR1 and mFOLR1, but the CAR candidates were also negatively selected against human and murine FOLR2 and FOLR4 variants, respectively. This selection for human and murine FOLR1-specific CAR candidates was critical since the members of the folate receptor family have a high degree of similarity [
35].
In contrast, the other reported fully human anti-FOLR1 scFv, C4, was generated using a guided selection approach [
56]. In this study, a human Fab fragment was isolated from a phage library using the light chain of the murine monoclonal antibody Mov19 as a template for pairing of human heavy or light chains, respectively, displayed on phages and an ovarian cell line overexpressing FOLR1. Whether the C4 Fab recognizes other FOLR1 variants in human and mouse is unclear.
For functional assessment of CAR molecules, we decided on a second-generation CAR architecture which comprises a CD8a spacer, CD8a transmembrane domain, and a 4-1BB co-stimulatory endodomain (signal 2 for T cell activation), followed by a CD3z-coding endodomain, to provide activation signal 1. We selected 4-1BB as a co-stimulatory domain since it has previously been described to promote proliferation and persistence of T cells [
57,
58,
59]. To assess the impact of individual scFv domains on CAR-mediated target cell lysis, cytokine release, and cell expansion, we compared all CAR constructs in parallel in repeated co-cultures in an automated screening assay. Co-culture assays of CAR T cells, with repetitive addition of target cells, were performed to identify CAR candidates with the best performance in vitro. It was proposed that multiple engagements into T cell effector function, upon repeated antigen exposure, may serve as an in vitro surrogate for the challenging situation CAR T cells may encounter in patients [
11,
12,
13,
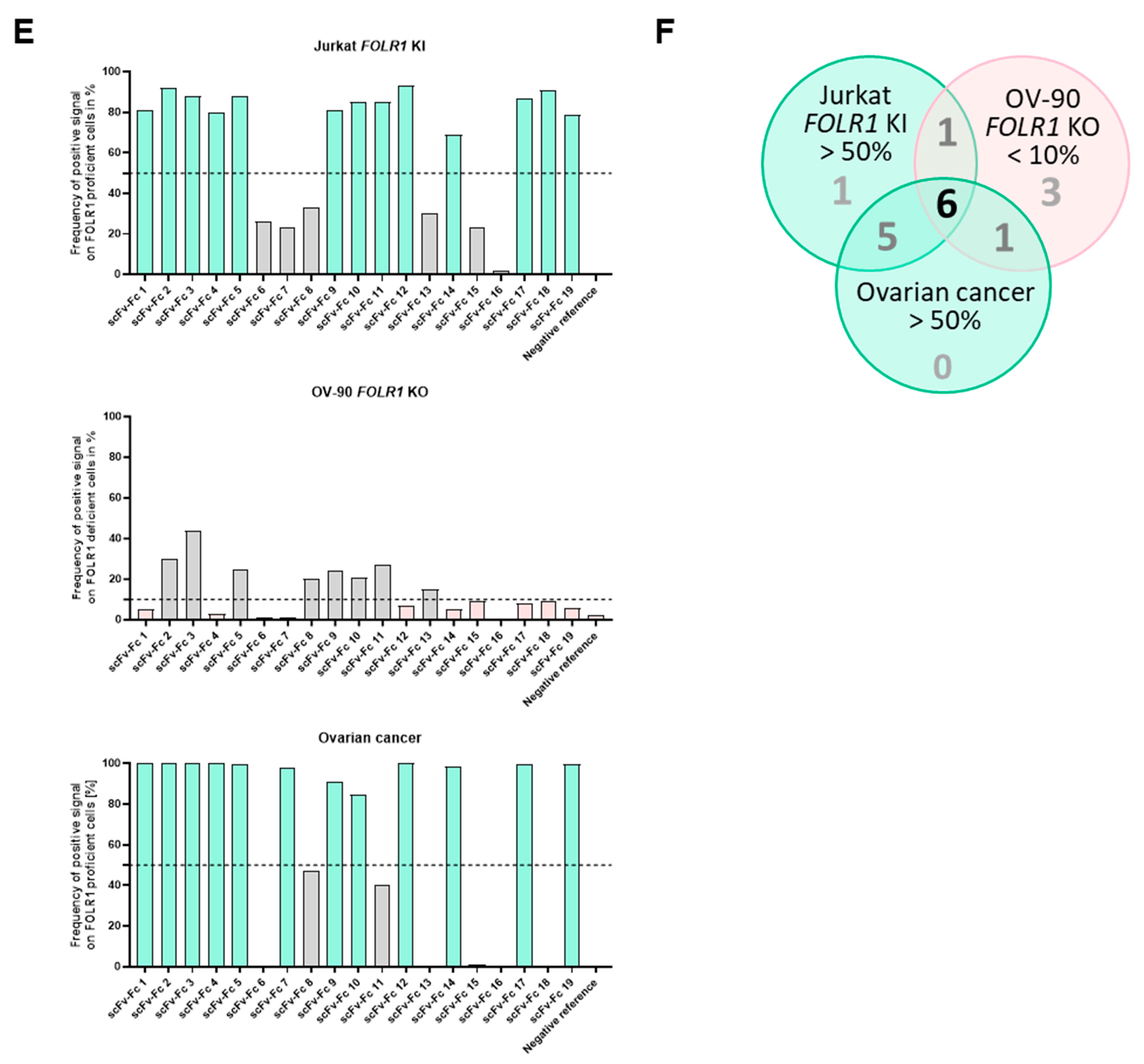
60]. Although the majority of anti-FOLR1 CAR candidates (17 out of 19) mediated killing of FOLR1-expressing target cells in the first round of co-culture, not all of these anti-FOLR1 CAR candidates (only 12) were able to induce target cell killing in the second and third round. One of these anti-FOLR1 CAR candidates also induced repeated killing of
FOLR1 KO target cells, emphasizing the importance of assessing antigen-independent activation early in the development process. Moreover, this indicates that specificity of a CAR T cell may differ from the specificity of an isolated scFv. This effect may be meditated by different avidities [
61]. While most anti-FOLR1 CAR candidates (10 from 12) were able to induce profound release of IFN-γ upon target cell encounter in the third round, only a few anti-FOLR1 CAR candidates (4 out of 12) expanded in all rounds and all three donors. Induction of CAR T cell proliferation underlined their functional superiority [
62]. These findings indicate that CAR T cell functionality changes after repeated contact with target cells, highlighting that advanced in vitro assays are critical for functional assessment of CAR T cells.
Subsequently, we performed additional in vitro assays to further characterize the anti-FOLR1 CAR T cell candidates under challenging conditions, i.e., repetitive killing assays at low effector-to-target cell ratios (1:2), and selected the best performing candidates.
So far, one other fully human anti-FOLR1 CAR, C4-27z CAR, was reported [
63]. The reported C4-27z CAR and our described fully human anti-FOLR1 CAR differ in the co-stimulatory domains, where the C4-27z CAR incorporates a CD27 domain and our fully human anti-FOLR1 CAR bears a 4-1BB endodomain. CD27 (also known as TNFRSF7) is a member of the TNFR superfamily, and it is expressed by immune cells [
63]. The authors hypothesized that CD27 co-stimulation may enhance expansion, effector function, and survival in vitro. Additionally, CD27 co-stimulation should support human CAR T-cell persistence and anti-tumor activity in vivo [
64]. However, a comparison of CD27 against CD28 or 4-1BB co-stimulation was not performed by the authors to show a benefit of CD27 signaling. Although the C4-27z CAR showed promising in vitro and in vivo data, it was not assessed in repetitive killing assays, which is an in vitro dysfunctional model [
65]. The fully human anti-FOLR1 CAR which we described was selected based on its functionality in this dysfunctionality model.
In addition to the extensive functional in vitro characterization of CAR T cell candidates, we set up a new tissue cross-reactivity workflow based on multiplexed immunofluorescence. We aimed to include on-target/off-tumor reactivity early in the selection process, since several clinical CAR T cell studies reported either off-target or on-target/off-tumor reactivity of CAR T cells [
22]. Examples for on-target/off-tumor reactivity are CAR T cells targeting ErbB2/her2 [
29], EGFR [
27], and CAIX-specific CAR T cells [
66]. By assessment of off-target or on-target/off-tumor reactivity early in the development process, we intended to focus on the most promising candidates and efficiently perform the subsequent development.
For evaluating off-tumor and off-target reactions specifically related to CAR T cell therapy, the FDA recommends several assays that are crucial for evaluating the safety and efficacy of CAR T cell products. Tissue cross-reactivity studies provide information from more complex tissue samples and help to identify potential off-target and off-tumor interactions that may lead to adverse effects in patients.
Spatial multiomics offers several advantages over traditional FDA-recommended assays for assessing off-target reactions for CAR T cells. Spatial multiomics techniques, such as spatial transcriptomics and spatial proteomics, provide detailed spatial information about gene expression, protein expression, and cell–cell interactions within tissue samples. This comprehensive spatial data allow for the identification of off-tumor and off-target interactions within the context of the tissue microenvironment, providing insights into potential off-tumor effects that may not be captured by traditional assays.
The tissue cross-reactivity workflow started with an on-target analysis of the CAR candidates. Cell lines with defined FOLR1 expression, i.e., knock-in or knock-out, respectively, were used to determine on-target reactivity. Here, we already excluded candidates which did not meet our criteria. Next, we analyzed the on-tumor binding profile of the anti-FOLR1 scFv-Fc fusion proteins using primary human ovarian cancer tissues.
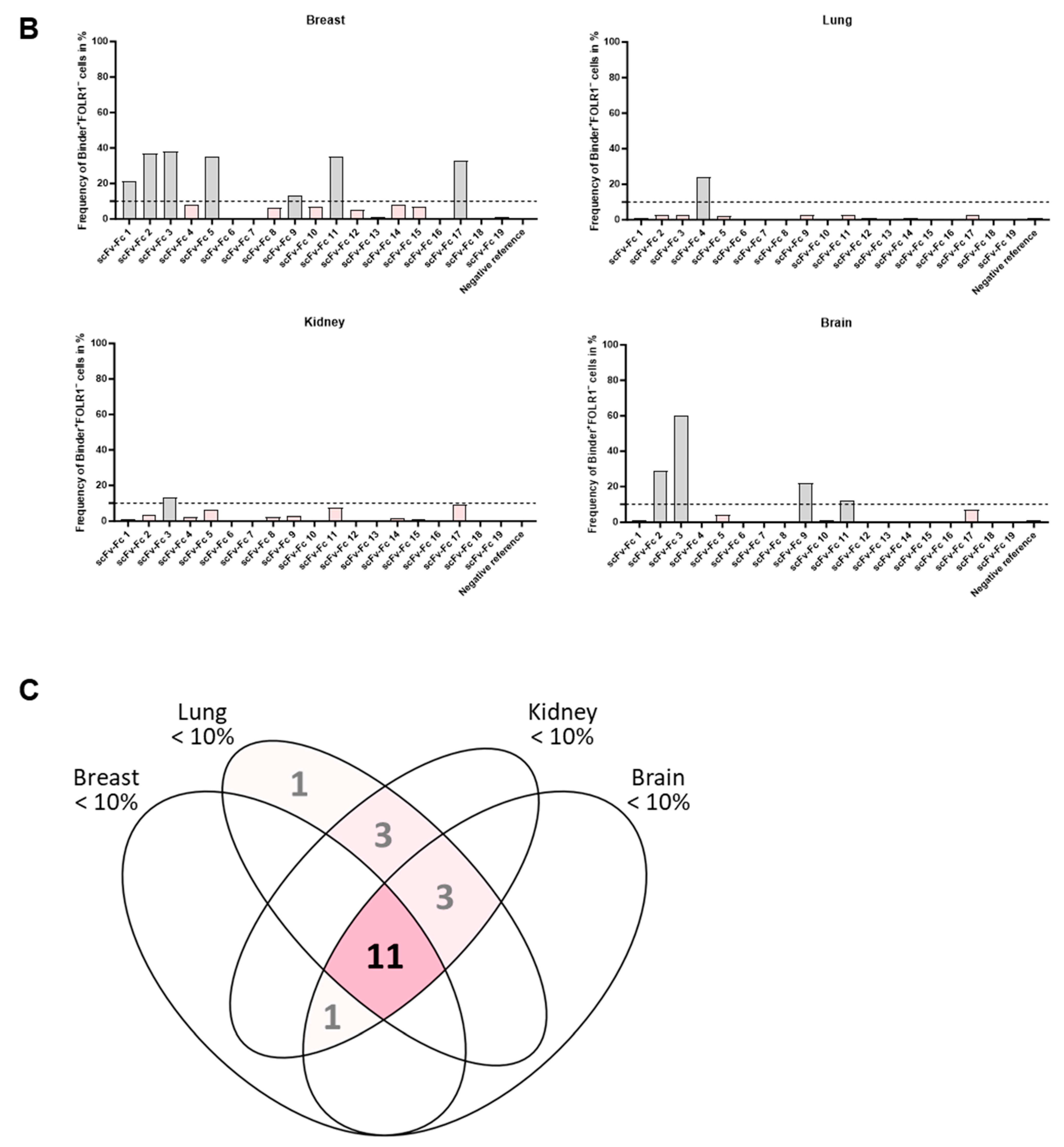
It was previously described that FOLR1 expression is not restricted to ovarian cancer but can also be found in healthy breast, kidney, and lung tissue. This indicates off-tumor expression of FOLR1. FOLR1 expression is locally restricted to the apical surface of epithelial cells, which may make the receptor potentially inaccessible to blood-circulating drugs [
36,
67]. Consequently, analysis of off-tumor binding of new CAR candidates targeting FOLR1 is important.
Subsequent analysis of the anti-FOLR1 scFv-Fc binder candidates on non-malignant tissues enabled further exclusion of candidates with potential off-target specificity. The analysis of tissue micro arrays allowed for the reduction of the number of potential candidates with lowest off-target specificity to three. Finally, we nominated a lead candidate for further development based on superior performance in comprehensive functionality assays as well as most promising safety profile.
In contrast, the other reported fully human anti-FOLR1 scFv, C4, was exclusively tested for on-tumor reactivity on ovarian carcinoma by immunohistochemistry [
56].
A limitation of the study is that the scFv-Fc fusion proteins may not fully reflect the interaction of a CAR T cell with its cognate antigen. The interaction of a CAR T cell with an antigen-expressing cell may be modulated by multiple interactions, giving rise to an avidity that cannot be resembled by a single molecule like a scFv-Fc.
Albeit, the FOLR1 expression in mice has not fully been investigated yet [
68], mouse–human cross-reactive binders—as characterized in this study—could be a valuable tool to address on-target, off-tumor expression of FOLR1 in murine tissue and potential on-target, off-tumor related toxicity in mice. Future studies should address whether FOLR1 expression in mice is comparable to human FOLR1 expression in terms of tissue distribution, cellular localization, and intensity. Subsequently, it could be assessed if cross-reactive scFvs bind to murine tissues expressing FOLR1 to foresee potential toxicities. Finally, such a hypothesis could be confirmed by applying murine-reactive CAR T cells to mice and carefully monitor animals for potential toxicities.
Besides a safety assessment in mice, future studies should address efficacy of the fully human anti-FOLR1 CAR in a relevant in vivo ovarian cancer model. These data will support the clinical translation of a fully human anti-FOLR1 CAR T cell therapy for a broad range of FOLR1-expressing epithelial cancers.
Taken together, we describe a novel workflow to identify and characterize fully human lead CAR candidates. Our approach combines a series of complementary assays including highly specific candidate selection from a human antibody phage library with positive and negative selection steps. Moreover, lead candidate selection was based on CAR functionality as well as on-target/off-tumor reactivity to mitigate potential toxicities. Thereby, CAR therapy may be improved by more efficient lead selection and reduced risk of potential toxicities in patients.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}