
Mechanisms of Insulin Signaling as a Potential Therapeutic Method in Intestinal Diseases


Abstract
1. Introduction
2. Mechanisms of Intestinal Inflammation
2.1. Factors Related to the Intestinal Microbiota That Affect the Maintenance of Homeostasis in the Intestinal Tract
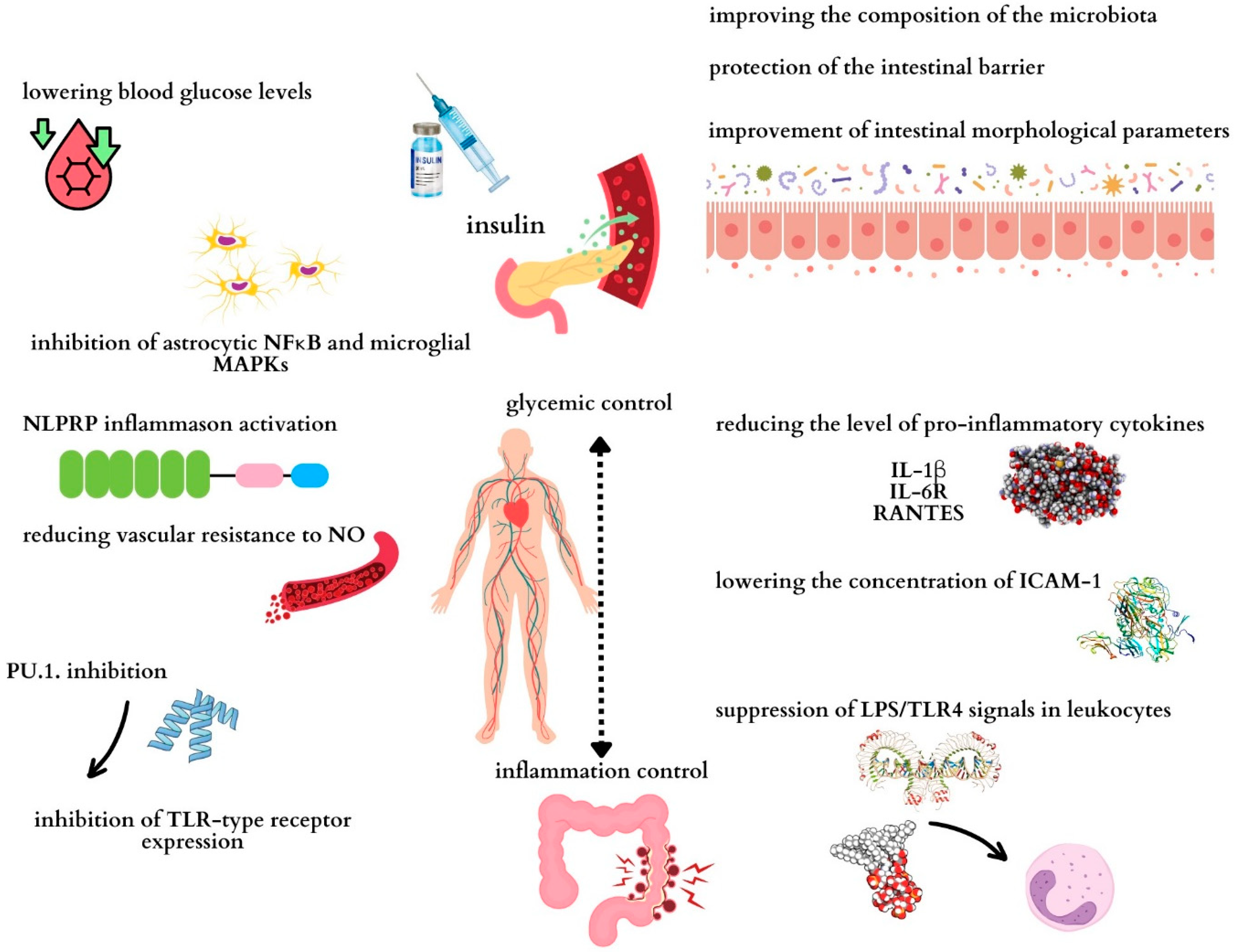
2.2. Receptor-Mediated Insulin Signaling
3. Regulation of Inflammation in the Intestines
3.1. The Role of Insulin in Regulating Inflammation
3.2. Reducing Inflammation in the Gut by Influencing the Gut Microbiota
3.3. Other Factors That Reduce Inflammation in the Gut
3.3.1. A High-Fiber Diet
3.3.2. Dietary Supplements
3.3.3. Physical Activity
3.3.4. Adequate Amount of Sleep
4. Effects of Insulin-like Growth Factor 1 (IGF-1) on Intestinal Inflammation and Gut Microbiota
5. Effects of Glucagon-like Peptide-1 (GLP-1) on Intestinal Inflammation and Gut Microbiota
6. Tumor Necrosis Factor-like Ligand 1A (TL1A)
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 74. [Google Scholar] [CrossRef]
- Petagna, L.; Antonelli, A.; Ganini, C.; Bellato, V.; Campanelli, M.; Divizia, A.; Efrati, C.; Franceschilli, M.; Guida, A.M.; Ingallinella, S.; et al. Pathophysiology of Crohn’s disease inflammation and recurrence. Biol. Direct 2020, 15, 23. [Google Scholar] [CrossRef]
- Du, L.; Ha, C. Epidemiology and Pathogenesis of Ulcerative Colitis. Gastroenterol. Clin. N. Am. 2020, 49, 643–654. [Google Scholar] [CrossRef]
- Windsor, J.W.; Kaplan, G.G. Evolving Epidemiology of IBD. Curr. Gastroenterol. Rep. 2019, 21, 40. [Google Scholar] [CrossRef]
- Li, T.; Han, B.; Wang, L.; Sun, L.; Cai, Y.; Yu, M.; Xiao, W.; Yang, H. Activation of mucosal insulin receptor exacerbates intestinal inflammation by promoting tissue resident memory T cells differentiation through EZH2. J. Transl. Med. 2024, 22, 78. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cui, W.; Li, X.; Yang, H. Interaction Between Commensal Bacteria, Immune Response and the Intestinal Barrier in Inflammatory Bowel Disease. Front. Immunol. 2021, 12, 761981. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, J.; Yan, W.; Xu, L. Macrophage polarization in inflammatory bowel disease. Cell Commun. Signal 2023, 21, 367. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Huang, J.; Ayansola, H.; Masatoshi, H.; Zhang, B. Intestinal Stem Cells and Immune Cell Relationships: Potential Therapeutic Targets for Inflammatory Bowel Diseases. Front. Immunol. 2021, 11, 623691. [Google Scholar] [CrossRef]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bris, R.; Saez, A.; Herrero-Fernandez, B.; Rius, C.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2696. [Google Scholar] [CrossRef]
- Jiang, P.; Zheng, C.; Xiang, Y.; Malik, S.; Su, D.; Xu, G.; Zhang, M. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor. Rev. 2023, 69, 28–42. [Google Scholar] [CrossRef]
- Wang, B.; Shen, J. NF-κB Inducing Kinase Regulates Intestinal Immunity and Homeostasis. Front. Immunol. 2022, 13, 895636. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, P.; Sassi, F. Gut Microbiota, Immune System, and Bone. Calcif. Tissue Int. 2018, 102, 415–425. [Google Scholar] [CrossRef]
- Guo, M.; Liu, H.; Yu, Y.; Zhu, X.; Xie, H.; Wei, C.; Mei, C.; Shi, Y.; Zhou, N.; Qin, K.; et al. Lactobacillus rhamnosus GG ameliorates osteoporosis in ovariectomized rats by regulating the Th17/Treg balance and gut microbiota structure. Gut Microbes 2023, 15, 2190304. [Google Scholar] [CrossRef]
- Zhu, P.; Lu, T.; Wu, J.; Fan, D.; Liu, B.; Zhu, X.; Guo, H.; Du, Y.; Liu, F.; Tian, Y.; et al. Gut microbiota drives macrophage-dependent self-renewal of intestinal stem cells via niche enteric serotonergic neurons. Cell Res. 2022, 32, 555–569. [Google Scholar] [CrossRef]
- Allam-Ndoul, B.; Castonguay-Paradis, S.; Veilleux, A. Gut Microbiota and Intestinal Trans-Epithelial Permeability. Int. J. Mol. Sci. 2020, 21, 6402. [Google Scholar] [CrossRef] [PubMed]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243, Erratum in Gut 2023, 72, e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cao, S.Q.; Lin, Z.M.; He, S.J.; Zuo, J.P. NOD-like receptors in autoimmune diseases. Acta Pharmacol. Sin. 2021, 42, 1742–1756. [Google Scholar] [CrossRef]
- Feng, S.; Zhang, C.; Chen, S.; He, R.; Chao, G.; Zhang, S. TLR5 Signaling in the Regulation of Intestinal Mucosal Immunity. J. Inflamm. Res. 2023, 16, 2491–2501. [Google Scholar] [CrossRef]
- Zhao, Z.; Ning, J.; Bao, X.Q.; Shang, M.; Ma, J.; Li, G.; Zhang, D. Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome 2021, 9, 226. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Jin, T.; Yi, C.; Ocansey, D.K.W.; Mao, F. The role of NOD2 in intestinal immune response and microbiota modulation: A therapeutic target in inflammatory bowel disease. Int. Immunopharmacol. 2022, 113, 109466. [Google Scholar] [CrossRef] [PubMed]
- DePeaux, K.; Delgoffe, G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021, 21, 785–797. [Google Scholar] [CrossRef]
- Andres, S.F.; Simmons, J.G.; Mah, A.T.; Santoro, M.A.; Van Landeghem, L.; Lund, P.K. Insulin receptor isoform switching in intestinal stem cells, progenitors, differentiated lineages and tumors: Evidence that IR-B limits proliferation. J. Cell Sci. 2013, 126, 5645–5656. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cell Metab. 2018, 28, 922–934. [Google Scholar] [CrossRef]
- Norton, L.; Shannon, C.; Gastaldelli, A.; DeFronzo, R.A. Insulin: The master regulator of glucose metabolism. Metabolism 2022, 129, 155142. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, W.; Wu, R.; Li, J.; Park, S.A.; Tu, E.; Zanvit, P.; Xu, J.; Liu, O.; Cain, A.; et al. High Glucose Intake Exacerbates Autoimmunity through Reactive-Oxygen-Species-Mediated TGF-β Cytokine Activation. Immunity 2019, 51, 671–681. [Google Scholar] [CrossRef]
- Yassin, M.; Sadowska, Z.; Tritsaris, K.; Kissow, H.; Hansen, C.H.F.; Forman, J.L.; Rogler, G.; Troelsen, J.T.; Pedersen, A.E.; Olsen, J. Rectal Insulin Instillation Inhibits Inflammation and Tumor Development in Chemically Induced Colitis. J. Crohn’s Colitis 2018, 12, 1459–1474. [Google Scholar] [CrossRef]
- Makhijani, P.; Basso, P.J.; Chan, Y.T.; Chen, N.; Baechle, J.; Khan, S.; Furman, D.; Tsai, S.; Winer, D.A. Regulation of the immune system by the insulin receptor in health and disease. Front. Endocrinol. 2023, 14, 1128622. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Matuschik, L.; Riabov, V.; Schmuttermaier, C.; Sevastyanova, T.; Weiss, C.; Klüter, H.; Kzhyshkowska, J. Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes. Int. J. Mol. Sci. 2022, 23, 1385. [Google Scholar] [CrossRef]
- Mouri, M.I.; Badireddy, M. Hyperglycemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430900/ (accessed on 24 April 2023).
- Chang, Y.W.; Hung, L.C.; Chen, Y.C.; Wang, W.H.; Lin, C.Y.; Tzeng, H.H.; Suen, J.L.; Chen, Y.H. Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front. Immunol. 2021, 11, 587229. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Changes in Cells Associated with Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 2397. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yun, J.S.; Ko, S.H. Advanced Glycation End Products and Their Effect on Vascular Complications in Type 2 Diabetes Mellitus. Nutrients 2022, 14, 3086. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.; George, T.P.; Mujammami, M.; Isnani, A.; Alfadda, A.A. The association of cell adhesion molecules and selectins (VCAM-1, ICAM-1, E-selectin, L-selectin, and P-selectin) with microvascular complications in patients with type 2 diabetes: A follow-up study. Front. Endocrinol. 2023, 14, 1072288. [Google Scholar] [CrossRef]
- Wronka, M.; Krzemińska, J.; Młynarska, E.; Rysz, J.; Franczyk, B. The Influence of Lifestyle and Treatment on Oxidative Stress and Inflammation in Diabetes. Int. J. Mol. Sci. 2022, 23, 5743. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Mohanty, P.; Deopurkar, R.; Sia, C.L.; Korzeniewski, K.; Abuaysheh, S.; Chaudhuri, A.; Dandona, P. Acute modulation of toll-like receptors by insulin. Diabetes Care 2008, 31, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Tryggestad, J.B.; Shah, R.D.; Braffett, B.H.; Bacha, F.; Gidding, S.S.; Gubitosi-Klug, R.A.; Shah, A.S.; Urbina, E.M.; Levitt Katz, L.E.; TODAY Study Group. Circulating adhesion molecules and associations with HbA1c, hypertension, nephropathy, and retinopathy in the Treatment Options for type 2 Diabetes in Adolescent and Youth study. Pediatr. Diabetes 2020, 21, 923–931. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Dai, P.; Liu, L.; Yang, Y.; Liu, X.; Li, Y.; Liao, Z. The effect of short-term intensive insulin therapy on inflammatory cytokines in patients with newly diagnosed type 2 diabetes. J. Diabetes 2022, 14, 192–204. [Google Scholar] [CrossRef]
- Singh, V.; Kaur, R.; Kumari, P.; Pasricha, C.; Singh, R. ICAM-1 and VCAM-1: Gatekeepers in various inflammatory and cardiovascular disorders. Clin. Chim. Acta 2023, 548, 117487. [Google Scholar] [CrossRef]
- Ruiz, H.H.; Nguyen, A.; Wang, C.; He, L.; Li, H.; Hallowell, P.; McNamara, C.; Schmidt, A.M. AGE/RAGE/DIAPH1 axis is associated with immunometabolic markers and risk of insulin resistance in subcutaneous but not omental adipose tissue in human obesity. Int. J. Obes. 2021, 45, 2083–2094. [Google Scholar] [CrossRef]
- Zhang, Z.; Amorosa, L.F.; Coyle, S.M.; Macor, M.A.; Birnbaum, M.J.; Lee, L.Y.; Haimovich, B. Insulin-Dependent Regulation of mTORC2-Akt-FoxO Suppresses TLR4 Signaling in Human Leukocytes: Relevance to Type 2 Diabetes. Diabetes 2016, 65, 2224–2234. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Kashfi, K.; Ghasemi, A. Vascular nitric oxide resistance in type 2 diabetes. Cell Death Dis. 2023, 14, 410. [Google Scholar] [CrossRef]
- Stettner, N.; Rosen, C.; Bernshtein, B.; Gur-Cohen, S.; Frug, J.; Silberman, A.; Sarver, A.; Carmel-Neiderman, N.N.; Eilam, R.; Biton, I.; et al. Induction of Nitric-Oxide Metabolism in Enterocytes Alleviates Colitis and Inflammation-Associated Colon Cancer. Cell Rep. 2018, 23, 1962–1976. [Google Scholar] [CrossRef]
- Huang, C.T.; Lue, J.H.; Cheng, T.H.; Tsai, Y.J. Glycemic control with insulin attenuates sepsis-associated encephalopathy by inhibiting glial activation via the suppression of the nuclear factor kappa B and mitogen-activated protein kinase signaling pathways in septic rats. Brain Res. 2020, 1738, 146822. [Google Scholar] [CrossRef] [PubMed]
- Darra, A.; Singh, V.; Jena, A.; Popli, P.; Nada, R.; Gupta, P.; Bhadada, S.K.; Singh, A.K.; Sharma, V.; Bhattacharya, A.; et al. Hyperglycemia is associated with duodenal dysbiosis and altered duodenal microenvironment. Sci. Rep. 2023, 13, 11038. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef]
- Dubois, N.; Muñoz-Garcia, J.; Heymann, D.; Renodon-Cornière, A. High glucose exposure drives intestinal barrier dysfunction by altering its morphological, structural and functional properties. Biochem. Pharmacol. 2023, 216, 115765. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef] [PubMed]
- Al Bataineh, M.T.; Künstner, A.; Dash, N.R.; Alsafar, H.S.; Ragab, M.; Schmelter, F.; Sina, C.; Busch, H.; Ibrahim, S.M. Uncovering the relationship between gut microbial dysbiosis, metabolomics, and dietary intake in type 2 diabetes mellitus and in healthy volunteers: A multi-omics analysis. Sci. Rep. 2023, 13, 17943. [Google Scholar] [CrossRef]
- He, J.L.; Zhao, Y.W.; Yang, J.L.; Ju, J.M.; Ye, B.Q.; Huang, J.Y.; Huang, Z.H.; Zhao, W.Y.; Zeng, W.F.; Xia, M.; et al. Enhanced interactions among gut mycobiomes with the deterioration of glycemic control. Med 2024, 18, 909–925.e7. [Google Scholar] [CrossRef]
- Abdellatif, A.M.; Sarvetnick, N.E. Current understanding of the role of gut dysbiosis in type 1 diabetes. J. Diabetes 2019, 11, 632–644. [Google Scholar] [CrossRef]
- Luo, M.; Sun, M.; Wang, T.; Zhang, S.; Song, X.; Liu, X.; Wei, J.; Chen, Q.; Zhong, T.; Qin, J. Gut microbiota and type 1 diabetes: A two-sample bidirectional Mendelian randomization study. Front. Cell Infect. Microbiol. 2023, 13, 1163898. [Google Scholar] [CrossRef]
- Wang, H.; Tang, W.; Zhang, P.; Zhang, Z.; He, J.; Zhu, D.; Bi, Y. Modulation of gut microbiota contributes to effects of intensive insulin therapy on intestinal morphological alteration in high-fat-diet-treated mice. Acta Diabetol. 2020, 57, 455–467. [Google Scholar] [CrossRef]
- Huang, L.; Sililas, P.; Thonusin, C.; Tongsong, T.; Luewan, S.; Chattipakorn, N.; Chattipakorn, S.C. Association Between Gut Microbiota and Insulin Therapy in Women With Gestational Diabetes Mellitus. Can. J. Diabetes 2022, 46, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, K.; Saha, S.; Umar, S. Health Benefits of Dietary Fiber for the Management of Inflammatory Bowel Disease. Biomedicines 2022, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, D.; Huang, J.; Zuo, T. The gut microbiome: Linking dietary fiber to inflammatory diseases. Med. Microecol. 2022, 14, 100070. [Google Scholar] [CrossRef]
- Recharla, N.; Geesala, R.; Shi, X.Z. Gut Microbial Metabolite Butyrate and Its Therapeutic Role in Inflammatory Bowel Disease: A Literature Review. Nutrients 2023, 15, 2275. [Google Scholar] [CrossRef] [PubMed]
- Rios-Morales, M.; Vieira-Lara, M.A.; Homan, E.; Homan, E.; Langelaar-Makkinje, M.; Gerding, A.; Li, Z.; Huijkman, N.; Rensen, P.C.N.; Wolters, J.C.; et al. Butyrate oxidation attenuates the butyrate-induced improvement of insulin sensitivity in myotubes. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166476. [Google Scholar] [CrossRef]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Chakraborty, P.; Gangopadhyay, M.; Sahu, R.; Medala, V.; John, A.; Reddy, P.H.; De Feo, V.; et al. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells. 2021, 10, 1340. [Google Scholar] [CrossRef]
- He, J.; Zhang, P.; Shen, L.; Niu, L.; Tan, Y.; Chen, L.; Zhao, Y.; Bai, L.; Hao, X.; Li, X.; et al. Short-Chain Fatty Acids and Their Association with Signaling Pathways in Inflammation, Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2020, 21, 6356. [Google Scholar] [CrossRef]
- Liu, J.L.; Segovia, I.; Yuan, X.L.; Gao, Z.H. Controversial Roles of Gut Microbiota-Derived Short-Chain Fatty Acids (SCFAs) on Pancreatic β-Cell Growth and Insulin Secretion. Int. J. Mol. Sci. 2020, 21, 910. [Google Scholar] [CrossRef]
- Kiani, A.K.; Bonetti, G.; Donato, K.; Bertelli, M. Dietary supplements for intestinal inflammation. J. Prev. Med. Hyg. 2022, 63, E214–E220. [Google Scholar] [CrossRef]
- Virk, M.S.; Virk, M.A.; He, Y.; Tufail, T.; Gul, M.; Qayum, A.; Rehman, A.; Rashid, A.; Ekumah, J.N.; Han, X.; et al. The Anti-Inflammatory and Curative Exponent of Probiotics: A Comprehensive and Authentic Ingredient for the Sustained Functioning of Major Human Organs. Nutrients 2024, 16, 546. [Google Scholar] [CrossRef]
- Liu, N.; Feng, G.; Zhang, X.; Hu, Q.; Sun, S.; Sun, J.; Sun, Y.; Wang, R.; Zhang, Y.; Wang, P.; et al. The Functional Role of Lactoferrin in Intestine Mucosal Immune System and Inflammatory Bowel Disease. Front. Nutr. 2021, 8, 759507. [Google Scholar] [CrossRef] [PubMed]
- D’Antongiovanni, V.; Pellegrini, C.; Antonioli, L.; Benvenuti, L.; Di Salvo, C.; Flori, L.; Piccarducci, R.; Daniele, S.; Martelli, A.; Calderone, V.; et al. Palmitoylethanolamide Counteracts Enteric Inflammation and Bowel Motor Dysfunctions in a Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 748021. [Google Scholar] [CrossRef] [PubMed]
- Kianifar, H.; Jafari, S.A.; Khalesi, M.; Kiani, M.; Sadeghi, T.; Jaafari, M.R.; Amani, F. The Impact of Silymarin on the Symptom Severity in Pediatric Patients with Inflammatory Bowel Disease: A Randomized Clinical Trial. Int. J. Pediatr. 2023, 11, 18274–18286. [Google Scholar] [CrossRef]
- Marton, L.T.; Goulart, R.A.; Carvalho, A.C.A.; Barbalho, S.M. Omega Fatty Acids and Inflammatory Bowel Diseases: An Overview. Int. J. Mol. Sci. 2019, 20, 4851. [Google Scholar] [CrossRef] [PubMed]
- Maioli, T.U.; Trindade, L.M.; Souza, A.; Torres, L.; Andrade, M.E.R.; Cardoso, V.N.; Generoso, S.V. Non-pharmacologic strategies for the management of intestinal inflammation. Biomed. Pharmacother. 2022, 145, 112414. [Google Scholar] [CrossRef]
- Tiong, H.T.; Fan, D.; Frampton, C.; Ananthakrishnan, A.N.; Gearry, R.B. Physical Activity Is Associated With A Decreased Risk Of Developing Inflammatory Bowel Disease: A Systematic Review And Meta-Analysis. J. Crohn’s Colitis 2024, 18, 1476–1485. [Google Scholar] [CrossRef]
- Protano, C.; Gallè, F.; Volpini, V.; De Giorgi, A.; Mazzeo, E.; Ubaldi, F.; Romano Spica, V.; Vitali, M.; Valeriani, F. Physical activity in the prevention and management of inflammatory bowel disease: A systematic review. J. Public Health 2024. [Google Scholar] [CrossRef]
- Davis, S.P.; Crane, P.B.; Bolin, L.P.; Johnson, L.A. An integrative review of physical activity in adults with inflammatory bowel disease. Intest. Res. 2022, 20, 43–52. [Google Scholar] [CrossRef]
- Qazi, T.; Farraye, F.A. Sleep and Inflammatory Bowel Disease: An Important Bi-Directional Relationship. Inflamm. Bowel Dis. 2019, 25, 843–852. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, W.H.; Li, S.X.; He, Z.M.; Zhu, W.L.; Ji, Y.B.; Wang, Z.; Zhu, X.M.; Yuan, K.; Bao, Y.P.; et al. Gut microbiota modulates the inflammatory response and cognitive impairment induced by sleep deprivation. Mol. Psychiatry 2021, 26, 6277–6292. [Google Scholar] [CrossRef]
- Bermingham, K.M.; Stensrud, S.; Asnicar, F.; Valdes, A.M.; Franks, P.W.; Wolf, J.; Hadjigeorgiou, G.; Davies, R.; Spector, T.D.; Segata, N.; et al. Exploring the relationship between social jetlag with gut microbial composition, diet and cardiometabolic health, in the ZOE PREDICT 1 cohort. Eur. J. Nutr. 2023, 62, 3135–3147. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle, J.F. Insulin-like growth factors in the gastrointestinal tract and liver. Endocrinol. Metab. Clin. N. Am. 2012, 41, 409–423. [Google Scholar] [CrossRef]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef]
- Schwarzer, M.; Makki, K.; Storelli, G.; Machuca-Gayet, I.; Srutkova, D.; Hermanova, P.; Martino, M.E.; Balmand, S.; Hudcovic, T.; Heddi, A.; et al. Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition. Science 2016, 351, 854–857. [Google Scholar] [CrossRef]
- Kareem, K.Y.; Loh, T.C.; Foo, H.L.; Akit, H.; Samsudin, A.A. Effects of dietary postbiotic and inulin on growth performance, IGF1 and GHR mRNA expression, faecal microbiota and volatile fatty acids in broilers. BMC Vet. Res. 2016, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- García Navas, P.; Ruíz Del Prado, M.Y.; Villoslada Blanco, P.; Recio Fernández, E.; Ruíz Del Campo, M.; Pérez Matute, P. Composition of the microbiota in patients with growth hormone deficiency before and after treatment with growth hormone. An. Pediatr. 2024, 100, 404–411. [Google Scholar] [CrossRef]
- Yan, J.; Herzog, J.W.; Tsang, K.; Brennan, C.A.; Bower, M.A.; Garrett, W.S.; Sartor, B.R.; Aliprantis, A.O.; Charles, J.F. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 2016, 113, E7554–E7563. [Google Scholar] [CrossRef]
- Chen, T.; Zheng, F.; Tao, J.; Tan, S.; Zeng, L.; Peng, X.; Wu, B. Insulin-Like Growth Factor-1 Contributes to Mucosal Repair by β-Arrestin2-Mediated Extracellular Signal-Related Kinase Signaling in Experimental Colitis. Am. J. Pathol. 2015, 185, 2441–2453. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Chen, J.; Chen, S.; Li, Z.; Liu, H.; Bai, Y.; Zhi, F. Embryonic stem cell-derived mesenchymal stem cells promote colon epithelial integrity and regeneration by elevating circulating IGF-1 in colitis mice. Theranostics 2020, 10, 12204–12222. [Google Scholar] [CrossRef]
- Chen, H.; Min, X.H.; Wang, Q.Y.; Leung, F.W.; Shi, L.; Zhou, Y.; Yu, T.; Wang, C.M.; An, G.; Sha, W.H.; et al. Pre-activation of mesenchymal stem cells with TNF-α, IL-1β and nitric oxide enhances its paracrine effects on radiation-induced intestinal injury. Sci. Rep. 2015, 5, 8718. [Google Scholar] [CrossRef]
- Cao, X.; Duan, L.; Hou, H.; Liu, Y.; Chen, S.; Zhang, S.; Liu, Y.; Wang, C.; Qi, X.; Liu, N.; et al. IGF-1C hydrogel improves the therapeutic effects of MSCs on colitis in mice through PGE2-mediated M2 macrophage polarization. Theranostics 2020, 10, 7697–7709. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Saito, H.; Fukushima, R.; Hashiguchi, Y.; Lin, M.T.; Inoue, T.; Fukatsu, K.; Muto, T.; Takenaka, A.; Takahashi, S.; et al. Effects of growth hormone and insulin-like growth factor 1 (IGF-1) treatments on the nitrogen metabolism and hepatic IGF-1-messenger RNA expression in postoperative parenterally fed rats. J. Parenter. Enteral Nutr. 1996, 20, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Soendergaard, C.; Kvist, P.H.; Thygesen, P.; Reslow, M.; Nielsen, O.H.; Kopchick, J.J.; Holm, T.L. Characterization of Growth Hormone Resistance in Experimental and Ulcerative Colitis. Int. J. Mol. Sci. 2017, 18, 2046. [Google Scholar] [CrossRef] [PubMed]
- Bohin, N.; McGowan, K.P.; Keeley, T.M.; Carlson, E.A.; Yan, K.S.; Samuelson, L.C. Insulin-like Growth Factor-1 and mTORC1 Signaling Promote the Intestinal Regenerative Response After Irradiation Injury. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 797–810. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell 2018, 23, 787–793. [Google Scholar] [CrossRef]
- Grønbek, H.; Thøgersen, T.; Frystyk, J.; Vilstrup, H.; Flyvbjerg, A.; Dahlerup, J.F. Low free and total insulinlike growth factor I (IGF-I) and IGF binding protein-3 levels in chronic inflammatory bowel disease: Partial normalization during prednisolone treatment. Am. J. Gastroenterol. 2002, 97, 673–678. [Google Scholar] [CrossRef]
- Sipos, F.; Galamb, O.; Herszényi, L.; Molnár, B.; Solymosi, N.; Zágoni, T.; Berczi, L.; Tulassay, Z. Elevated insulin-like growth factor 1 receptor, hepatocyte growth factor receptor and telomerase protein expression in mild ulcerative colitis. Scand. J. Gastroenterol. 2008, 43, 289–298. [Google Scholar] [CrossRef]
- Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Kuśnierz-Cabala, B.; Bonior, J.; Jaworek, J.; Ambroży, T.; Gil, K.; Olszanecki, R.; et al. Essential Role of Growth Hormone and IGF-1 in Therapeutic Effect of Ghrelin in the Course of Acetic Acid-Induced Colitis. Int. J. Mol. Sci. 2017, 18, 1118. [Google Scholar] [CrossRef]
- Grau-Bové, C.; González-Quilen, C.; Cantini, G.; Nardini, P.; Espina, B.; Bani, D.; Terra, X.; Blay, M.; Rodríguez-Gallego, E.; Luconi, M.; et al. GLP1 Exerts Paracrine Activity in the Intestinal Lumen of Human Colon. Int. J. Mol. Sci. 2022, 23, 3523. [Google Scholar] [CrossRef]
- Anbazhagan, A.N.; Thaqi, M.; Priyamvada, S.; Jayawardena, D.; Kumar, A.; Gujral, T.; Chatterjee, I.; Mugarza, E.; Saksena, S.; Onyuksel, H.; et al. GLP-1 nanomedicine alleviates gut inflammation. Nanomedicine 2017, 13, 659–665. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, C.; Zhang, H.; Li, L.; Fan, T.; Jin, Z. The alleviating effect and mechanism of GLP-1 on ulcerative colitis. Aging 2023, 15, 8044–8060. [Google Scholar] [CrossRef] [PubMed]
- Funayama, T.; Nozu, T.; Ishioh, M.; Igarashi, S.; Sumi, C.; Saito, T.; Toki, Y.; Hatayama, M.; Yamamoto, M.; Shindo, M.; et al. Centrally administered GLP-1 analogue improves intestinal barrier function through the brain orexin and the vagal pathway in rats. Brain Res. 2023, 1809, 148371. [Google Scholar] [CrossRef] [PubMed]
- Abdalqadir, N.; Adeli, K. GLP-1 and GLP-2 Orchestrate Intestine Integrity, Gut Microbiota, and Immune System Crosstalk Microorganisms 2022, 10, 2061. Microorganisms 2022, 10, 2061. [Google Scholar] [CrossRef]
- Bang-Berthelsen, C.H.; Holm, T.L.; Pyke, C.; Simonsen, L.; Søkilde, R.; Pociot, F.; Heller, R.S.; Folkersen, L.; Kvist, P.H.; Jackerott, M.; et al. GLP-1 Induces Barrier Protective Expression in Brunner’s Glands and Regulates Colonic Inflammation. Inflamm. Bowel Dis. 2016, 22, 2078–2097. [Google Scholar] [CrossRef]
- Ebbesen, M.; Kissow, H.; Hartmann, B.; Kielsen, K.; Sørensen, K.; Stinson, S.E.; Frithioff-Bøjsøe, C.; Esmann Fonvig, C.; Holm, J.C.; Hansen, T.; et al. Glucagon-Like Peptide-1 Is Associated With Systemic Inflammation in Pediatric Patients Treated With Hematopoietic Stem Cell Transplantation. Front. Immunol. 2021, 12, 793588. [Google Scholar] [CrossRef]
- Su, Y.; Liu, N.; Zhang, Z.; Li, H.; Ma, J.; Yuan, Y.; Shi, M.; Liu, J.; Zhao, Z.; Zhang, Z.; et al. Cholecystokinin and glucagon-like peptide-1 analogues regulate intestinal tight junction, inflammation, dopaminergic neurons and α-synuclein accumulation in the colon of two Parkinson’s disease mouse models. Eur. J. Pharmacol. 2022, 926, 175029. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Koufakis, T.; Popovic, D.; Maltese, G.; Mustafa, O.; Doumas, M.; Giouleme, O.; Kotsa, K.; Germanidis, G. GLP-1 Receptor Agonists in Obese Patients with Inflammatory Bowel Disease: From Molecular Mechanisms to Clinical Considerations and Practical Recommendations for Safe and Effective Use. Curr. Obes. Rep. 2023, 12, 61–74. [Google Scholar] [CrossRef]
- Yue, W.; Li, Y.; Ou, D.; Yang, Q. The GLP-1 receptor agonist liraglutide protects against oxidized LDL-induced endothelial inflammation and dysfunction via KLF2. Life 2019, 71, 1347–1354. [Google Scholar] [CrossRef]
- Mehdi, S.F.; Pusapati, S.; Anwar, M.S.; Lohana, D.; Kumar, P.; Nandula, S.A.; Nawaz, F.K.; Tracey, K.; Yang, H.; LeRoith, D.; et al. Glucagon-like peptide-1: A multi-faceted anti-inflammatory agent. Front. Immunol. 2023, 14, 1148209. [Google Scholar] [CrossRef]
- Amato, A.; Mulè, F. Neural Regeneration Research. Mumbai 2019, 11, 1901–1902. [Google Scholar] [CrossRef]
- Zou, Z.; Wang, Z. Liraglutide attenuates intestinal ischemia/reperfusion injury via NF-κB and PI3K/Akt pathways in mice. Life Sci. 2022, 309, 121045. [Google Scholar] [CrossRef] [PubMed]
- Insuela, D.B.R.; Carvalho, V.F. Glucagon and glucagon-like peptide-1 as novel anti-inflammatory and immunomodulatory compounds. Eur. J. Pharmacol. 2017, 812, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Cani, P.D. Gut microbiota and GLP-1. Rev. Endocr. Metab. Disord. 2014, 15, 189–196. [Google Scholar] [CrossRef]
- Angelini, G.; Russo, S.; Mingrone, G. Incretin hormones, obesity and gut microbiota. Peptides 2024, 178, 171216. [Google Scholar] [CrossRef]
- Salvi, P.S.; Cowles, R.A. Butyrate and the Intestinal Epithelium: Modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef]
- Xu, W.D.; Li, R.; Huang, A.F. Role of TL1A in Inflammatory Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 2022, 13, 891328. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, M.; Hokari, R. New and Emerging Treatments for Inflammatory Bowel Disease. Digestion 2023, 104, 74–81. [Google Scholar] [CrossRef]
- Siakavellas, S.I.; Sfikakis, P.P.; Bamias, G. The TL1A/DR3/DcR3 pathway in autoimmune rheumatic diseases. Semin. Arthritis Rheum. 2015, 45, 1–8. [Google Scholar] [CrossRef]
- Furfaro, F.; Alfarone, L.; Gilardi, D.; Correale, C.; Allocca, M.; Fiorino, G.; Argollo, M.; Zilli, A.; Zacharopoulou, E.; Loy, L.; et al. TL1A: A New Potential Target in the Treatment of Inflammatory Bowel Disease. Curr. Drug Targets 2021, 22, 760–769. [Google Scholar] [CrossRef]
- Danese, S.; Klopocka, M.; Scherl, E.J.; Romatowski, J.; Allegretti, J.R.; Peeva, E.; Vincent, M.S.; Schoenbeck, U.; Ye, Z.; Hassan-Zahraee, M.; et al. Anti-TL1A Antibody PF-06480605 Safety and Efficacy for Ulcerative Colitis: A Phase 2a Single-Arm Study. Clin. Gastroenterol. Hepatol. 2021, 19, 2324–2332.e6. [Google Scholar] [CrossRef]
- Clarke, A.W.; Poulton, L.; Shim, D.; Mabon, D.; Butt, D.; Pollard, M.; Pande, V.; Husten, J.; Lyons, J.; Tian, C.; et al. An anti-TL1A antibody for the treatment of asthma and inflammatory bowel disease. mAbs 2018, 10, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Valatas, V.; Kolios, G.; Bamias, G. TL1A (TNFSF15) and DR3 (TNFRSF25): A Co-stimulatory System of Cytokines With Diverse Functions in Gut Mucosal Immunity. Front. Immunol. 2019, 10, 583. [Google Scholar] [CrossRef]
- Jacob, N.; Kumagai, K.; Abraham, J.P.; Shimodaira, Y.; Ye, Y.; Luu, J.; Blackwood, A.Y.; Castanon, S.L.; Stamps, D.T.; Thomas, L.S.; et al. Direct signaling of TL1A-DR3 on fibroblasts induces intestinal fibrosis in vivo. Sci. Rep. 2020, 10, 18189. [Google Scholar] [CrossRef] [PubMed]
- Zwolak, A.; Chan, S.R.; Harvilla, P.; Mahady, S.; Armstrong, A.A.; Luistro, L.; Tamot, N.; Yamada, D.; Derebe, M.; Pomerantz, S.; et al. A stable, engineered TL1A ligand co-stimulates T cells via specific binding to DR3. Sci. Rep. 2022, 12, 20538. [Google Scholar] [CrossRef] [PubMed]
- Jacob, N.; Jacobs, J.P.; Kumagai, K.; Ha, C.W.Y.; Kanazawa, Y.; Lagishetty, V.; Altmayer, K.; Hamill, A.M.; Von Arx, A.; Sartor, R.B.; et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. 2018, 11, 1466–1476. [Google Scholar] [CrossRef]
- Bamias, G. At the Junction of Immunity and Barrier Function: The Immunomodulatory Protein TL1A May Also Regulate Intestinal Permeability. Dig. Dis. Sci. 2019, 64, 1728–1730. [Google Scholar] [CrossRef]
- Li, H.; Song, J.; Niu, G.; Zhang, H.; Guo, J.; Shih, D.Q.; Targan, S.R.; Zhang, X. TL1A blocking ameliorates intestinal fibrosis in the T cell transfer model of chronic colitis in mice. Pathol. Res. Pract. 2018, 214, 217–227. [Google Scholar] [CrossRef]
- Yang, M.; Jia, W.; Wang, D.; Han, F.; Niu, W.; Zhang, H.; Shih, D.Q.; Zhang, X. Effects and Mechanism of Constitutive TL1A Expression on Intestinal Mucosal Barrier in DSS-Induced Colitis. Dig. Dis. Sci. 2019, 64, 1844–1856. [Google Scholar] [CrossRef]
- Li, J.; Shi, W.; Sun, H.; Ji, Y.; Chen, Y.; Guo, X.; Sheng, H.; Shu, J.; Zhou, L.; Cai, T.; et al. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat. Commun. 2019, 10, 3371. [Google Scholar] [CrossRef]

{kind=link}
| Dietary Supplements | Effect on the Intestines |
|---|---|
| Probiotics |
|
| Butyrate | |
| Lactoferrin |
|
| Palmitoylethanolamide |
|
| Phosphatidecholine |
|
| Silymarin | |
| Omega-3 acid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarmakiewicz-Czaja, S.; Sokal-Dembowska, A.; Ferenc, K.; Filip, R. Mechanisms of Insulin Signaling as a Potential Therapeutic Method in Intestinal Diseases. Cells 2024, 13, 1879. https://doi.org/10.3390/cells13221879
Jarmakiewicz-Czaja S, Sokal-Dembowska A, Ferenc K, Filip R. Mechanisms of Insulin Signaling as a Potential Therapeutic Method in Intestinal Diseases. Cells. 2024; 13(22):1879. https://doi.org/10.3390/cells13221879
Chicago/Turabian StyleJarmakiewicz-Czaja, Sara, Aneta Sokal-Dembowska, Katarzyna Ferenc, and Rafał Filip. 2024. "Mechanisms of Insulin Signaling as a Potential Therapeutic Method in Intestinal Diseases" Cells 13, no. 22: 1879. https://doi.org/10.3390/cells13221879
APA StyleJarmakiewicz-Czaja, S., Sokal-Dembowska, A., Ferenc, K., & Filip, R. (2024). Mechanisms of Insulin Signaling as a Potential Therapeutic Method in Intestinal Diseases. Cells, 13(22), 1879. https://doi.org/10.3390/cells13221879