
Iron Regulates Cellular Proliferation by Enhancing the Expression of Glucose Transporter GLUT3 in the Liver

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture and Treatments
2.3. siRNA Transfection
2.4. RNA Isolation, cDNA Synthesis, and Real-Time PCR
2.5. Protein Extraction and Immunoblotting
2.6. Immunoprecipitation
2.7. Immunostaining
2.8. Cell Proliferation Assays
2.9. Statistical Analysis
3. Results
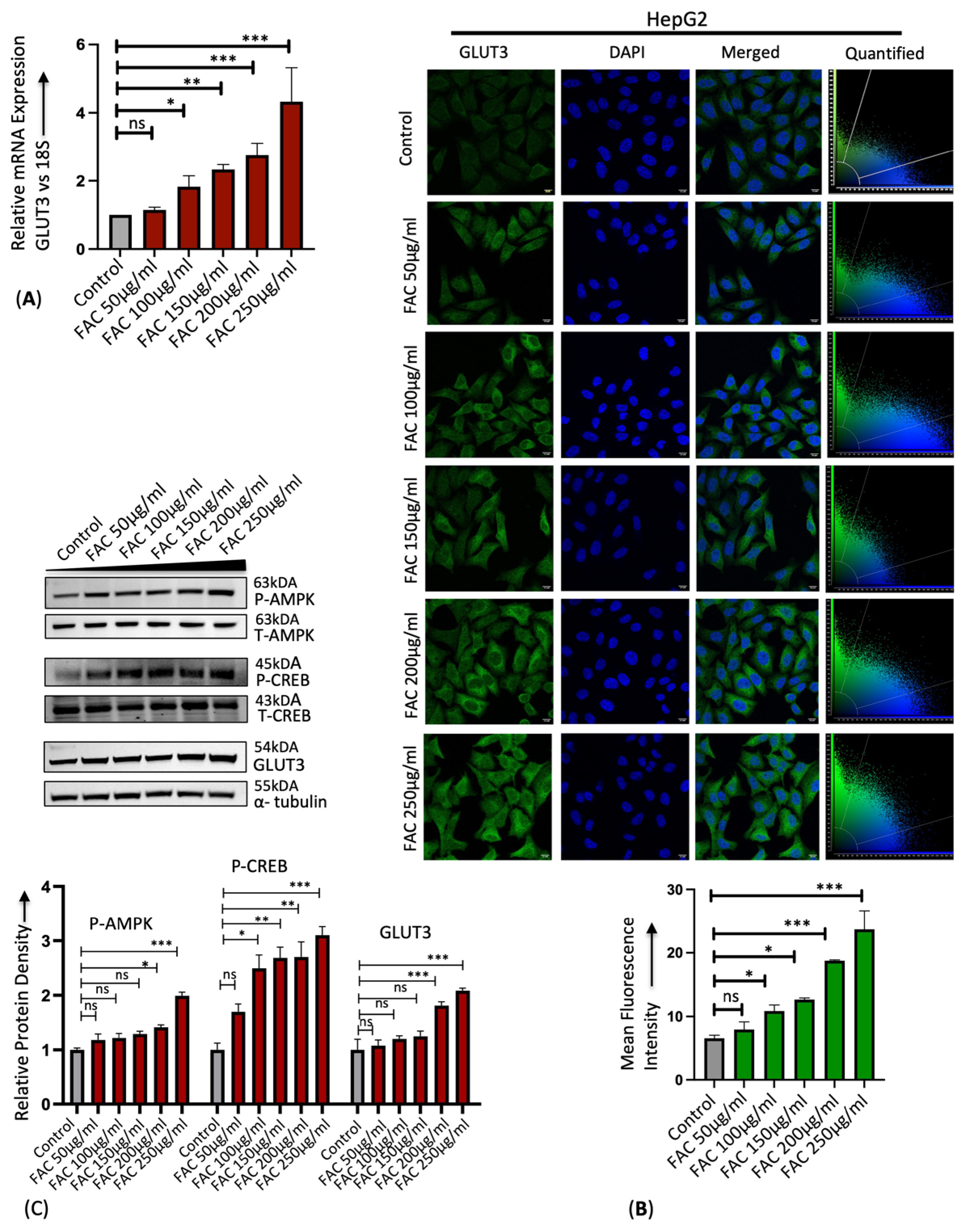
3.1. Iron Upregulates Hepatic GLUT3 Expression
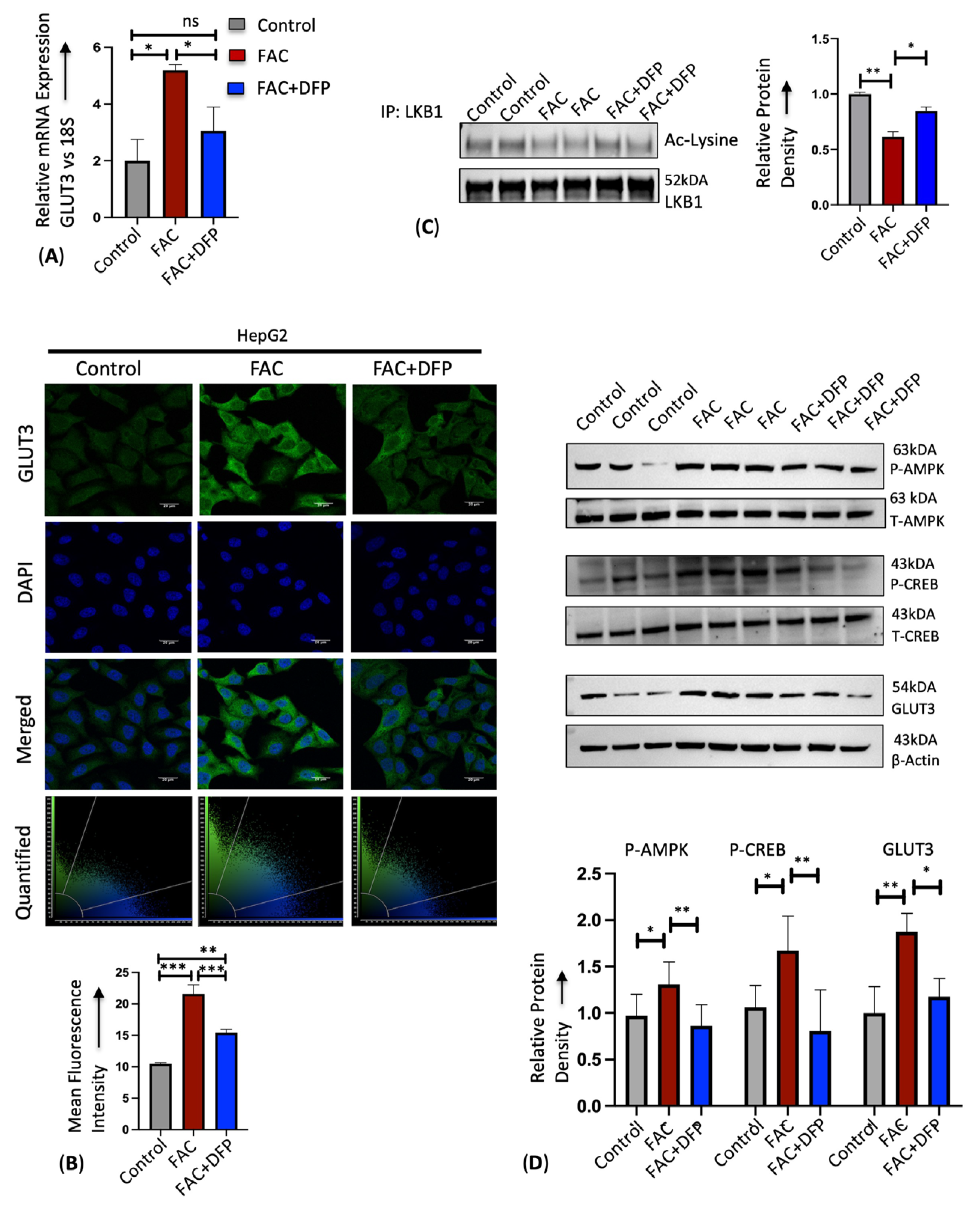
3.2. Iron Enhances GLUT3 Expression through LKB1/AMPK/CREB1 Signaling
3.3. GLUT3 Silencing Attenuates Iron-Mediated Cellular Hyperproliferation
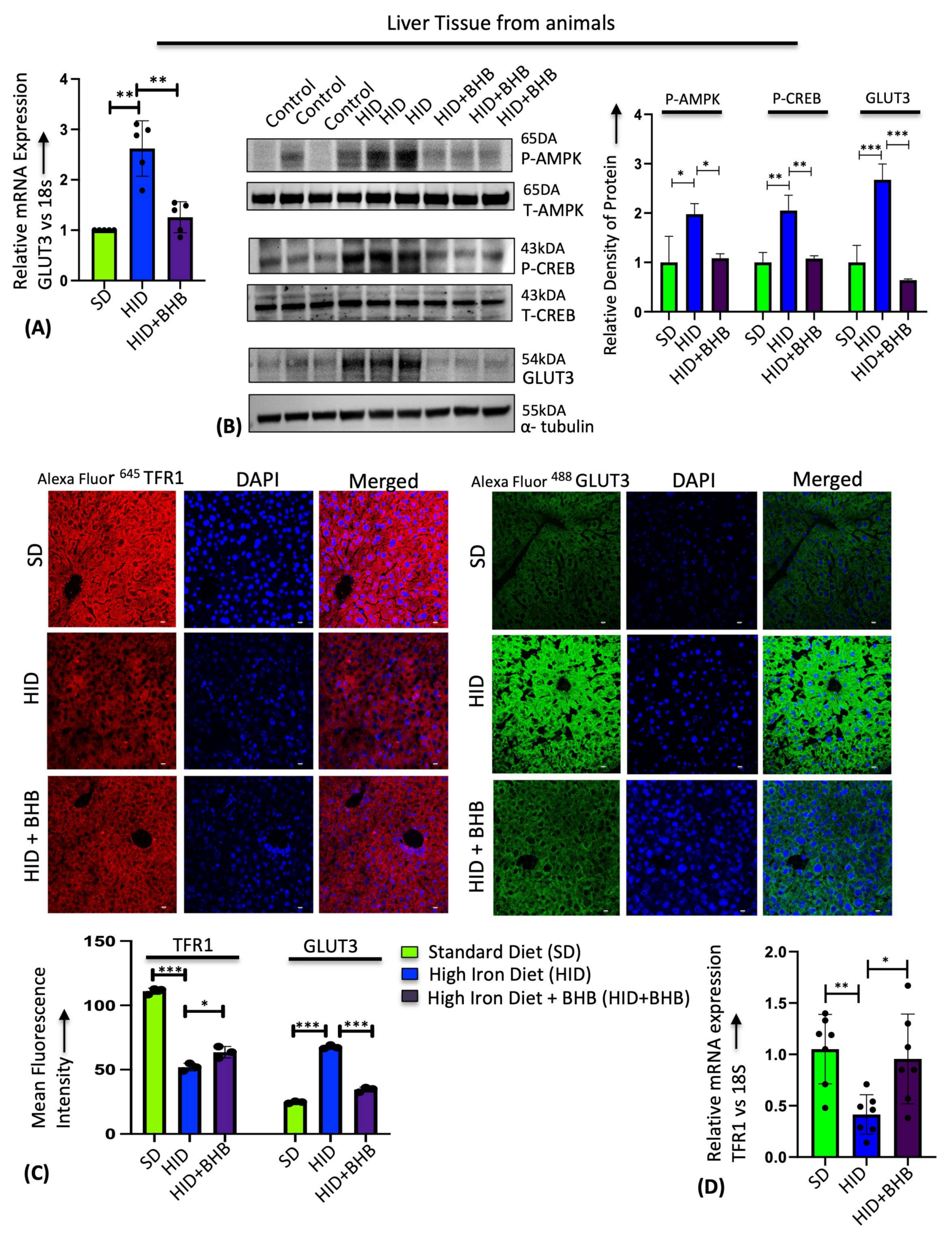
3.4. Sodium β-Hydroxybutyrate (BHB) Prevents Iron-Mediated GLUT3 Upregulation
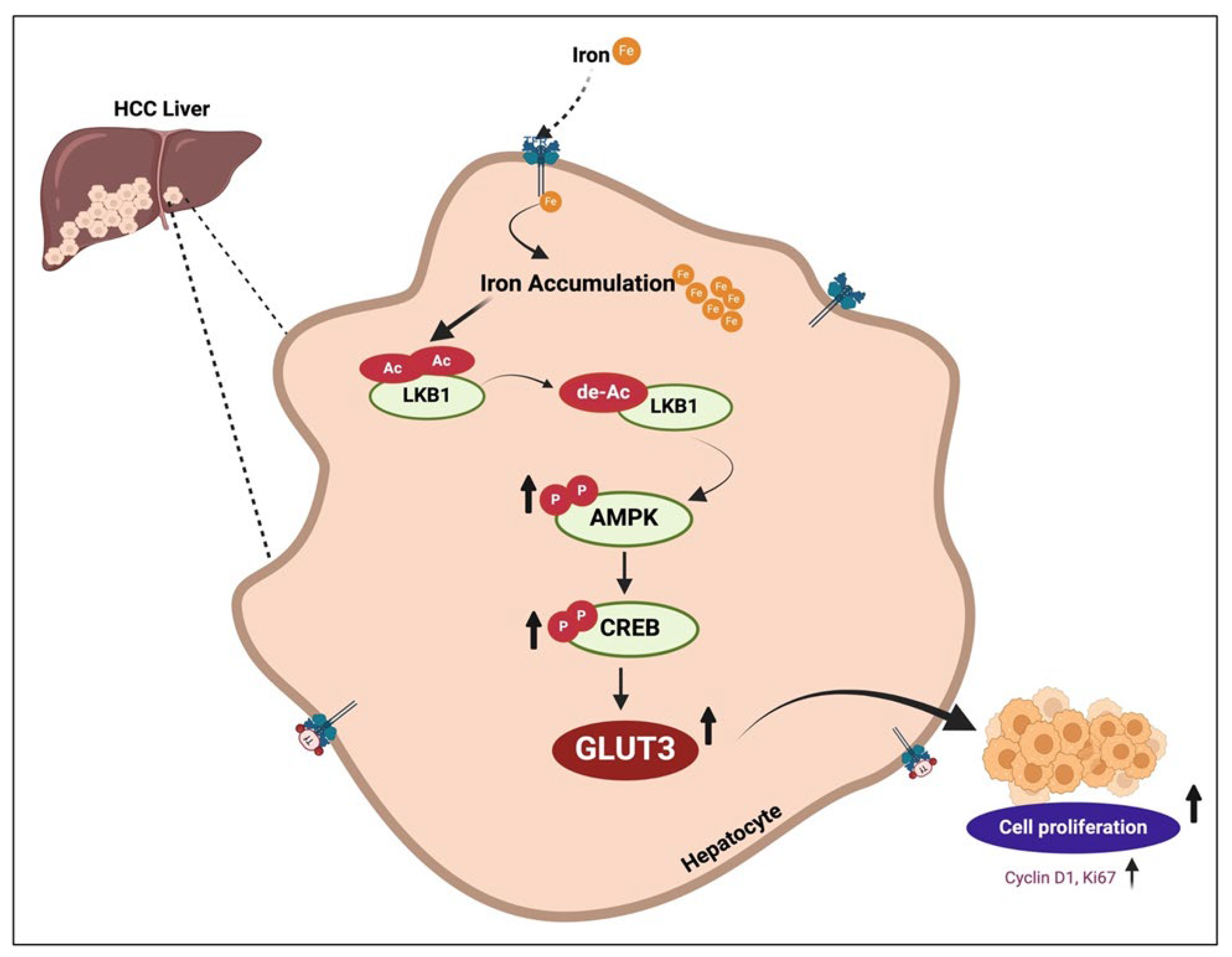
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Simcox, J.A.; McClain, D.A. Iron and diabetes risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Jahng, J.W.S.; Alsaadi, R.M.; Palanivel, R.; Song, E.; Hipolito, V.E.B.; Sung, H.K.; Botelho, R.J.; Russell, R.C.; Sweeney, G. Iron overload inhibits late stage autophagic flux leading to insulin resistance. EMBO Rep. 2019, 20, e47911. [Google Scholar] [CrossRef]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in the small intestine in health and disease. Pflugers Arch. 2020, 472, 1207–1248. [Google Scholar] [CrossRef] [PubMed]
- Kellett, G.L.; Brot-Laroche, E.; Mace, O.J.; Leturque, A. Sugar absorption in the intestine: The role of GLUT2. Annu. Rev. Nutr. 2008, 28, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Vulturar, R.; Chiș, A.; Pintilie, S.; Farcaș, I.M.; Botezatu, A.; Login, C.C.; Sitar-Taut, A.V.; Orasan, O.H.; Stan, A.; Lazea, C.; et al. One Molecule for Mental Nourishment and More: Glucose Transporter Type 1-Biology and Deficiency Syndrome. Biomedicines 2022, 10, 1249. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Cheng, Z.Q.; Brown, D.; Lodish, H.F. Liver glucose transporter: A basolateral protein in hepatocytes and intestine and kidney cells. Am. J. Physiol. 1990, 259 Pt 1, C279–C285. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Chen, X.; Bergmeier, S.C. Novel inhibitors of basal glucose transport as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Shriwas, P.; Roberts, D.; Li, Y.; Wang, L.; Qian, Y.; Bergmeier, S.; Hines, J.; Adhicary, S.; Nielsen, C.; Chen, X. A small-molecule pan-class I glucose transporter inhibitor reduces cancer cell proliferation in vitro and tumor growth in vivo by targeting glucose-based metabolism. Cancer Metab. 2021, 9, 14. [Google Scholar] [CrossRef]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650–654. [Google Scholar] [CrossRef]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef]
- Xia, H.; Chen, J.; Gao, H.; Kong, S.N.; Deivasigamani, A.; Shi, M.; Xie, T.; Hui, K.M. Hypoxia-induced modulation of glucose transporter expression impacts 18F-fluorodeoxyglucose PET-CT imaging in hepatocellular carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 787–797. [Google Scholar] [CrossRef]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Stoeltzing, O.; Weiss, T.S.; Schoelmerich, J.; Bosserhoff, A.K.; Hellerbrand, C. GLUT1 and GLUT3 expression are increased in hepatocellular carcinoma and promote tumorigenesis. Z. Gastroenterol. 2009, 47, P3_01. [Google Scholar] [CrossRef]
- Mandala, A.; Chen, W.J.; Armstrong, A.; Malhotra, M.R.; Chavalmane, S.; McCommis, K.S.; Chen, A.; Carpenter, D.; Biswas, P.; Gnana-Prakasam, J.P. PPARα agonist fenofibrate attenuates iron-induced liver injury in mice by modulating the Sirt3 and β-catenin signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G262–G269. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Choragiewicz, T.J.; Rejdak, R.; Fiedorowicz, M.; Turski, W.A.; Tulidowicz-Bielak, M.; Zrenner, E.; Schuettauf, F.; Zarnowski, T. Neuroprotection by acetoacetate and β-hydroxybutyrate against NMDA-induced RGC damage in rat—Possible involvement of kynurenic acid. Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Barisani, D.; Meneveri, R.; Ginelli, E.; Cassani, C.; Conte, D. Iron overload and gene expression in HepG2 cells: Analysis by differential display. FEBS Lett. 2000, 469, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Liu, C.M.; Guo, J.; Marcondes, A.M.; Deeg, J.; Li, X.; Guan, F. Iron overload induced by ferric ammonium citrate triggers reactive oxygen species-mediated apoptosis via both extrinsic and intrinsic pathways in human hepatic cells. Hum. Exp. Toxicol. 2016, 35, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Mandala, A.; Armstrong, A.; Girresch, B.; Zhu, J.; Chilakala, A.; Chavalmane, S.; Chaudhary, K.; Biswas, P.; Ogilvie, J.; Gnana-Prakasam, J.P. Fenofibrate prevents iron induced activation of canonical Wnt/β-catenin and oxidative stress signaling in the retina. NPJ Aging Mech. Dis. 2020, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.K.; Kim, D.H.; Bang, E.; Choi, Y.J.; Chung, H.Y. β-Hydroxybutyrate Suppresses Lipid Accumulation in Aged Liver through GPR109A-mediated Signaling. Aging Dis. 2020, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, R.; Kawano, Y.; Minami, A.; Nishiumi, S.; Yano, Y.; Yoshida, M.; Kodama, Y. β-hydroxybutyrate protects hepatocytes against endoplasmic reticulum stress in a sirtuin 1-independent manner. Arch. Biochem. Biophys. 2019, 663, 220–227. [Google Scholar] [CrossRef]
- Chaudhary, K.; Shinde, R.; Liu, H.; Gnana-Prakasam, J.P.; Veeranan-Karmegam, R.; Huang, L.; Ravishankar, B.; Bradley, J.; Kvirkvelia, N.; McMenamin, M.; et al. Amino acid metabolism inhibits antibody-driven kidney injury by inducing autophagy. J. Immunol. 2015, 194, 5713–5724. [Google Scholar] [CrossRef]
- Huang, J.; Simcox, J.; Mitchell, T.C.; Jones, D.; Cox, J.; Luo, B.; Cooksey, R.C.; Boros, L.G.; McClain, D.A. Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013, 27, 2845–2854. [Google Scholar] [CrossRef]
- Chaudhary, K.; Promsote, W.; Ananth, S.; Veeranan-Karmegam, R.; Tawfik, A.; Arjunan, P.; Martin, P.; Smith, S.B.; Thangaraju, M.; Kisselev, O.; et al. Iron Overload Accelerates the Progression of Diabetic Retinopathy in Association with Increased Retinal Renin Expression. Sci. Rep. 2018, 8, 3025. [Google Scholar] [CrossRef]
- Gnana-Prakasam, J.P.; Thangaraju, M.; Liu, K.; Ha, Y.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: Role of cystine/glutamate exchanger. Biochem. J. 2009, 424, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Carly, E.H.; Thorstensen, K.; Chua, A.C.G.; Graham, R.M.; Leedman, P.; Olynyk, J.K.; Trinder, D. The role of transferrin receptor 1 and 2 in transferrin-bound iron uptake in human hepatoma cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1567–C1575. [Google Scholar]
- Dai, W.; Xu, Y.; Mo, S.; Li, Q.; Yu, J.; Wang, R.; Ma, Y.; Ni, Y.; Xiang, W.; Han, L.; et al. GLUT3 induced by AMPK/CREB1 axis is key for withstanding energy stress and augments the efficacy of current colorectal cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 177. [Google Scholar] [CrossRef]
- Fargion, S.; Valenti, L.; Fracanzani, A.L. Role of iron in hepatocellular carcinoma. Clin. Liver Dis. 2014, 3, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Asare, G.A.; Mossanda, K.S.; Kew, M.C.; Paterson, A.C.; Kahler-Venter, C.P.; Siziba, K. Hepatocellular carcinoma caused by iron overload: A possible mechanism of direct hepatocarcinogenicity. Toxicology 2006, 219, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, R.; Lechel, A.; Vujic Spasic, M. Iron at the Interface of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 4097. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Pfeifhofer-Obermair, C.; Tymoszuk, P.; Petzer, V.; Weiss, G.; Nairz, M. Iron in the Tumor Microenvironment-Connecting the Dots. Front. Oncol. 2018, 8, 549. [Google Scholar] [CrossRef]
- Szymonik, J.; Wala, K.; Górnicki, T.; Saczko, J.; Pencakowski, B.; Kulbacka, J. The Impact of Iron Chelators on the Biology of Cancer Stem Cells. Int. J. Mol. Sci. 2021, 23, 89. [Google Scholar] [CrossRef]
- Brahim, O.; O’Sullivan, J. Iron chelators in cancer therapy. Biometals 2020, 33, 201–215. [Google Scholar] [CrossRef] [PubMed]
- King, K.L.; Hwang, J.J.; Chau, G.Y.; Tsay, S.H.; Chi, C.W.; Lee, T.G.; Wu, L.H.; Wu, C.W.; Lui, W.Y. Ki-67 expression as a prognostic marker in patients with hepatocellular carcinoma. J. Gastroenterol. Hepatol. 1998, 13, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Deane, N.G.; Parker, M.A.; Aramandla, R.; Diehl, L.; Lee, W.J.; Washington, M.K.; Nanney, L.B.; Shyr, Y.; Beauchamp, R.D. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 2001, 61, 5389–5395. [Google Scholar] [PubMed]
- Dmitrieva-Posocco, O.; Wong, A.C.; Lundgren, P.; Golos, A.M.; Descamps, H.C.; Dohnalová, L.; Cramer, Z.; Tian, Y.; Yueh, B.; Eskiocak, O.; et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature 2022, 605, 160–165. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Arnold, P.; Seyfried, T.N.; D’Agostino, D.P. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer 2014, 135, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Talib, W.H.; Mahmod, A.I.; Kamal, A.; Rashid, H.M.; Alashqar, A.M.D.; Khater, S.; Jamal, D.; Waly, M. Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Curr. Issues Mol. Biol. 2021, 43, 558–589. [Google Scholar] [CrossRef] [PubMed]
- Fidler, M.M.; Bray, F.; Soerjomataram, I. The global cancer burden and human development: A review. Scand. J. Public Health 2018, 46, 27–36. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron Metabolism in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2257. [Google Scholar] [CrossRef]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef]
- Chen, J.; Chloupková, M. Abnormal iron uptake and liver cancer. Cancer Biol. Ther. 2009, 8, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ferrara, N. Iron Metabolism in the Tumor Microenvironment: Contributions of Innate Immune Cells. Front. Immunol. 2021, 11, 626812. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Elliott, R.L.; Head, J.F. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. 2010, 30, 759–765. [Google Scholar]
- Boult, J.; Roberts, K.; Brookes, M.J.; Hughes, S.; Bury, J.P.; Cross, S.S.; Anderson, G.J.; Spychal, R.; Iqbal, T.; Tselepis, C. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin. Cancer Res. 2008, 14, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Brookes, M.J.; Hughes, S.; Turner, F.E.; Reynolds, G.; Sharma, N.; Ismail, T.; Berx, G.; McKie, A.T.; Hotchin, N.; Anderson, G.J.; et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006, 55, 1449–1460. [Google Scholar] [CrossRef]
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Hann, H.W.; Stahlhut, M.W.; Blumberg, B.S. Iron nutrition and tumor growth: Decreased tumor growth in iron-deficient mice. Cancer Res. 1988, 48, 4168–4170. [Google Scholar] [PubMed]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: Results from a randomized trial. J. Natl. Cancer Inst. 2008, 100, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Kalinowski, D.S.; Lau, S.; Jansson, P.J.; Lovejoy, D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta 2009, 1790, 702–717. [Google Scholar] [CrossRef]
- Jian, J.; Yang, Q.; Shao, Y.; Axelrod, D.; Smith, J.; Singh, B.; Krauter, S.; Chiriboga, L.; Yang, Z.; Li, J.; et al. A link between premenopausal iron deficiency and breast cancer malignancy. BMC Cancer 2013, 13, 307. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, W.; Abdul Razak, S.R.; Han, T.; Ahmad, N.H.; Li, X. Ferroptosis as a potential target for cancer therapy. Cell Death Dis 2023, 14, 460. [Google Scholar] [CrossRef] [PubMed]
- Bauckman, K.; Haller, E.; Taran, N.; Rockfield, S.; Ruiz-Rivera, A.; Nanjundan, M. Iron alters cell survival in a mitochondria-dependent pathway in ovarian cancer cells. Biochem. J. 2015, 466, 401–413. [Google Scholar] [CrossRef]
- Lv, X.; Lv, Y.; Dai, X. Lactate, histone lactylation and cancer hallmarks. Expert Rev. Mol. Med. 2023, 25, e7. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.C.; Sung, C.J.; Hsu, J.L.; Leu, W.J.; Guh, J.H.; Kung, F.L.; Hsu, L.C. The Combination of a Novel GLUT1 Inhibitor and Cisplatin Synergistically Inhibits Breast Cancer Cell Growth by Enhancing the DNA Damaging Effect and Modulating the Akt/mTOR and MAPK Signaling Pathways. Front Pharmacol. 2022, 13, 879748. [Google Scholar] [CrossRef]
- Pantaleon, M.; Harvey, M.B.; Pascoe, W.S.; James, D.E.; Kaye, P.L. Glucose transporter GLUT3: Ontogeny, targeting, and role in the mouse blastocyst. Proc. Natl. Acad. Sci. USA 1997, 94, 3795–3800. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.H.; Yang, C.C.; Kou, T.C.; Yang, C.E.; Dai, J.Z.; Chen, C.L.; Lin, C.W. Overexpression of GLUT3 promotes metastasis of triple-negative breast cancer by modulating the inflammatory tumor microenvironment. J. Cell. Physiol. 2021, 236, 4669–4680. [Google Scholar] [CrossRef]
- Yang, H.; Yang, S.; He, J.; Li, W.; Zhang, A.; Li, N.; Zhou, G.; Sun, B. Glucose transporter 3 (GLUT3) promotes lactylation modifications by regulating lactate dehydrogenase A (LDHA) in gastric cancer. Cancer Cell Int. 2023, 23, 303. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Ludovica Fracanzani, A.; Gatti, S.; Cairo, G.; Fargion, S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef]
- Carlos, A.R.; Weis, S.; Soares, M.P. Cross-Talk Between Iron and Glucose Metabolism in the Establishment of Disease Tolerance. Front. Immunol. 2018, 9, 2498. [Google Scholar] [CrossRef]
- Backe, M.B.; Moen, I.W.; Ellervik, C.; Hansen, J.B.; Mandrup-Poulsen, T. Iron Regulation of Pancreatic Beta-Cell Functions and Oxidative Stress. Annu. Rev. Nutr. 2016, 36, 241–273. [Google Scholar] [CrossRef]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef]
- Wu, G.Y.; Thompson, J.R. The effect of ketone bodies on alanine and glutamine metabolism in isolated skeletal muscle from the fasted chick. Biochem. J. 1988, 255, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Luo, W.; Wu, S.; Zhang, F.; Chen, X.; Ma, Y.; Mo, Y. Decreased expression of 3-hydroxybutyrate dehydrogenase 1 is a prognostic marker and promotes tumor progression in hepatocellular carcinoma. Pathol. Res. Pract. 2022, 238, 154111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Liu, Y.; Yang, D.; Jiao, Y.; Liu, Y. Expression and clinical significance of BDH1 in liver cancer. Medicine 2021, 100, e28013. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Tang, S.; Zhao, J.; Hao, Y.; Zhao, L.; Han, X.; Wang, X.; Zhang, L.; Li, R.; Zhou, X. β-Hydroxybutyrate Protects Against Cisplatin-Induced Renal Damage via Regulating Ferroptosis. Ren. Fail. 2024, 46, 2354918. [Google Scholar] [CrossRef]
- Qin, Y.; Bai, D.; Tang, M.; Zhang, M.; Zhao, L.; Li, J.; Yang, R.; Jiang, G. Ketogenic diet alleviates brain iron deposition and cognitive dysfunction via Nrf2-mediated ferroptosis pathway in APP/PS1 mouse. Brain Res. 2023, 1812, 148404. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, K.S.; Karmakar, E.; Park, C.; Garg, R.; Kung, G.P.; Kadakia, I.; Gopianand, J.S.; Arun, T.; Kisselev, O.; Gnana-Prakasam, J.P. Iron Regulates Cellular Proliferation by Enhancing the Expression of Glucose Transporter GLUT3 in the Liver. Cells 2024, 13, 1147. https://doi.org/10.3390/cells13131147
Ribeiro KS, Karmakar E, Park C, Garg R, Kung GP, Kadakia I, Gopianand JS, Arun T, Kisselev O, Gnana-Prakasam JP. Iron Regulates Cellular Proliferation by Enhancing the Expression of Glucose Transporter GLUT3 in the Liver. Cells. 2024; 13(13):1147. https://doi.org/10.3390/cells13131147
Chicago/Turabian StyleRibeiro, Kleber S., Eshani Karmakar, Christine Park, Richa Garg, George P. Kung, Isha Kadakia, Jyotsna S. Gopianand, Tejas Arun, Oleg Kisselev, and Jaya P. Gnana-Prakasam. 2024. "Iron Regulates Cellular Proliferation by Enhancing the Expression of Glucose Transporter GLUT3 in the Liver" Cells 13, no. 13: 1147. https://doi.org/10.3390/cells13131147
APA StyleRibeiro, K. S., Karmakar, E., Park, C., Garg, R., Kung, G. P., Kadakia, I., Gopianand, J. S., Arun, T., Kisselev, O., & Gnana-Prakasam, J. P. (2024). Iron Regulates Cellular Proliferation by Enhancing the Expression of Glucose Transporter GLUT3 in the Liver. Cells, 13(13), 1147. https://doi.org/10.3390/cells13131147