
Heavy Metal Interactions with Neuroglia and Gut Microbiota: Implications for Huntington’s Disease





{kind=link}
Abstract
1. Introduction
2. HD Pathophysiology
3. Current and Prospective Treatments
4. Glial Cells—HD
4.1. Microglia—HD
4.2. Astroglia (Astrocytes)—HD
4.3. Oligodendrocytes—HD
4.4. Synantocytes (NG2 Cells)—HD
5. Gut Microbiota
6. Gut–Brain Axis
7. Heavy Metals
7.1. Iron (Fe)—HD
7.2. Manganese (Mn)—HD
7.3. Copper (Cu)—HD
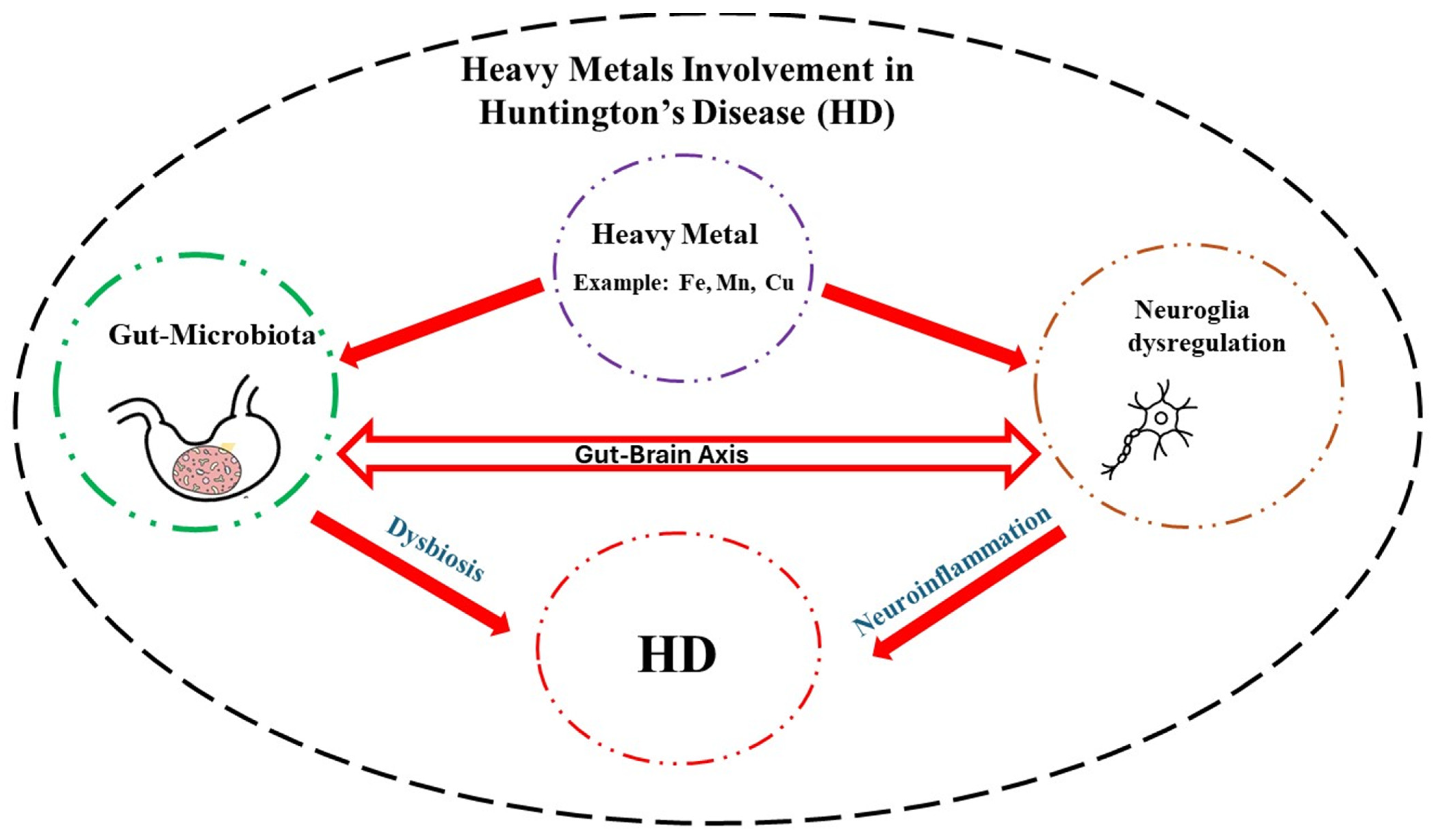
8. Heavy Metals and GM—HD
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ajitkumar, A.; De Jesus, O. Huntington Disease; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- D’Egidio, F.; Castelli, V.; Lombardozzi, G.; Ammannito, F.; Cimini, A.; d’Angelo, M. Therapeutic Advances in Neural Regeneration for Huntington’s Disease. Neural Regen. Res. 2024, 19, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, A.-W.; Roos, R.A.C. Aspiration Pneumonia and Death in Huntington’s Disease. PLoS Curr. 2012, 4, RRN1293. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.W.; Smeeth, L. The Prevalence of Huntington’s Disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Barron, J.C.; Hurley, E.P.; Parsons, M.P. Huntingtin and the Synapse. Front. Cell. Neurosci. 2021, 15, 689332. [Google Scholar] [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef]
- Khoshnan, A. Gut Microbiota as a Modifier of Huntington’s Disease Pathogenesis. J. Huntingt. Dis. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Estevam, B.; Matos, C.A.; Nóbrega, C. PolyQ Database—An Integrated Database on Polyglutamine Diseases. Database 2023, 2023, baad060. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Wright, G.E.; Hayden, M.R. Huntington Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- van der Plas, E.; Schultz, J.L.; Nopoulos, P.C. The Neurodevelopmental Hypothesis of Huntington’s Disease. J. Huntingt. Dis. 2020, 9, 217–229. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The Choreography of Neuroinflammation in Huntington’s Disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Donley, D.W.; Realing, M.; Gigley, J.P.; Fox, J.H. Iron Activates Microglia and Directly Stimulates Indoleamine-2,3-Dioxygenase Activity in the N171-82Q Mouse Model of Huntington’s Disease. PLoS ONE 2021, 16, e0250606. [Google Scholar] [CrossRef] [PubMed]
- Lobato, A.G.; Ortiz-Vega, N.; Zhu, Y.; Neupane, D.; Meier, K.K.; Zhai, R.G. Copper Enhances Aggregational Toxicity of Mutant Huntingtin in a Drosophila Model of Huntington’s Disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2024, 1870, 166928. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and Possible Future Therapeutic Options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517. [Google Scholar] [CrossRef] [PubMed]
- Paleacu, D. Tetrabenazine in the Treatment of Huntington’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 545–551. [Google Scholar] [PubMed]
- Sheridan, C. Questions Swirl around Failures of Disease-Modifying Huntington’s Drugs. Nat. Biotechnol. 2021, 39, 650–652. [Google Scholar] [CrossRef]
- Alkanli, S.S.; Alkanli, N.; Ay, A.; Albeniz, I. CRISPR/Cas9 Mediated Therapeutic Approach in Huntington’s Disease. Mol. Neurobiol. 2023, 60, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Estevez-Fraga, C.; van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential Disease-Modifying Therapies for Huntington’s Disease: Lessons Learned and Future Opportunities. Lancet Neurol. 2022, 21, 645–658. [Google Scholar] [CrossRef]
- Rook, M.E.; Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs 2022, 36, 105–119. [Google Scholar] [CrossRef]
- Yu, C.; Li, C.H.; Chen, S.; Yoo, H.; Qin, X.; Park, H. Decreased BDNF Release in Cortical Neurons of a Knock-in Mouse Model of Huntington’s Disease. Sci. Rep. 2018, 8, 16976. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Functional Analysis of Brain Derived Neurotrophic Factor (BDNF) in Huntington’s Disease. Aging 2021, 13, 6103–6114. [Google Scholar] [CrossRef] [PubMed]
- Speidell, A.; Bin Abid, N.; Yano, H. Brain-Derived Neurotrophic Factor Dysregulation as an Essential Pathological Feature in Huntington’s Disease: Mechanisms and Potential Therapeutics. Biomedicines 2023, 11, 2275. [Google Scholar] [CrossRef]
- Plinta, K.; Plewka, A.; Pawlicki, K.; Zmarzły, N.; Wójcik-Pędziwiatr, M.; Rudziński, M.; Krzak-Kubica, A.; Doręgowska-Stachera, M.; Rudzińska-Bar, M. The Utility of BDNF Detection in Assessing Severity of Huntington’s Disease. J. Clin. Med. 2021, 10, 5181. [Google Scholar] [CrossRef] [PubMed]
- Khan, E.; Mishra, S.K.; Mishra, R.; Mishra, A.; Kumar, A. Discovery of a Potent Small Molecule Inhibiting Huntington’s Disease (HD) Pathogenesis via Targeting CAG Repeats RNA and Poly Q Protein. Sci. Rep. 2019, 9, 16872. [Google Scholar] [CrossRef]
- Jarosińska, O.D.; Rüdiger, S.G.D. Molecular Strategies to Target Protein Aggregation in Huntington’s Disease. Front. Mol. Biosci. 2021, 8, 769184. [Google Scholar] [CrossRef]
- Raymond, L.A. Excitotoxicity in Huntington Disease. Clin. Neurosci. Res. 2003, 3, 121–128. [Google Scholar] [CrossRef]
- Kang, R.; Wang, L.; Sanders, S.S.; Zuo, K.; Hayden, M.R.; Raymond, L.A. Altered Regulation of Striatal Neuronal N-Methyl-D-Aspartate Receptor Trafficking by Palmitoylation in Huntington Disease Mouse Model. Front. Synaptic Neurosci. 2019, 11, 3. [Google Scholar] [CrossRef]
- Gao, J.; Wang, H.; Liu, Y.; Li, Y.-Y.; Chen, C.; Liu, L.-M.; Wu, Y.-M.; Li, S.; Yang, C. Glutamate and GABA Imbalance Promotes Neuronal Apoptosis in Hippocampus after Stress. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 499–512. [Google Scholar] [CrossRef]
- Saigoh, K.; Hirano, M.; Mitsui, Y.; Oda, I.; Ikegawa, A.; Samukawa, M.; Yoshikawa, K.; Yamagishi, Y.; Kusunoki, S.; Nagai, Y. Memantine Administration Prevented Chorea Movement in Huntington’s Disease: A Case Report. J. Med. Case Rep. 2023, 17, 431. [Google Scholar] [CrossRef] [PubMed]
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s Disease: A Randomized, Controlled Trial Using the NMDA-Antagonist Amantadine. Neurology 2002, 59, 694–699. [Google Scholar] [CrossRef]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; Difiglia, M. Early and Progressive Accumulation of Reactive Microglia in the Huntington Disease Brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef]
- Vonsattel, J.P.G.; Keller, C.; Pilar Amaya, M.D. Neuropathology of Huntington’s Disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 89, pp. 599–618. ISBN 978-0-444-51898-9. [Google Scholar]
- Tizabi, Y.; Getachew, B.; Hauser, S.R.; Tsytsarev, V.; Manhães, A.C.; Da Silva, V.D.A. Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction. Brain Sci. 2024, 14, 558. [Google Scholar] [CrossRef] [PubMed]
- Sanadgol, N. Editorial: Glial Cells as an Emerging Therapeutic Target in the Pathobiology of Central Nervous System Disorders: Friend or Foe? Front. Cell. Neurosci. 2023, 17, 1191743. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Couselo, F.L.; Bruno, J.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Neuroinflammation in Huntington’s Disease: A Starring Role for Astrocyteand Microglia. Curr. Neuropharmacol. 2022, 20, 1116–1143. [Google Scholar] [CrossRef]
- Carvalho, F.V.; Landis, H.E.; Getachew, B.; Diogenes Amaral Silva, V.; Ribeiro, P.R.; Aschner, M.; Tizabi, Y. Iron Toxicity, Ferroptosis and Microbiota in Parkinson’s Disease: Implications for Novel Targets. In Advances in Neurotoxicology; Elsevier: Amsterdam, The Netherlands, 2024; Volume 11, pp. 105–132. ISBN 978-0-443-21560-5. [Google Scholar]
- Pathak, D.; Sriram, K. Neuron-Astrocyte Omnidirectional Signaling in Neurological Health and Disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The Role of Microglia in the Development of Neurodegeneration. Neurol. Sci. 2020, 41, 3609–3615. [Google Scholar] [CrossRef]
- Soares, É.N.; Costa, A.C.D.S.; Ferrolho, G.D.J.; Ureshino, R.P.; Getachew, B.; Costa, S.L.; Da Silva, V.D.A.; Tizabi, Y. Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease. Cells 2024, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhu, W.; Liao, X.; Liu, W.; Hou, Y.; Wan, J. Expression of Toll-like Receptors in the Cerebellum during Pathogenesis of Prion Disease. Front. Behav. Neurosci. 2024, 18, 1341901. [Google Scholar] [CrossRef]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Microglia as Therapeutic Target in Central Nervous System Disorders. J. Pharmacol. Sci. 2020, 144, 102–118. [Google Scholar] [CrossRef]
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The Role of Toll-like Receptors and Neuroinflammation in Parkinson’s Disease. J. Neuroinflamm. 2022, 19, 135. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hernández, M.I.; Acosta-Saavedra, L.C.; Hernández-Kelly, L.C.; Loaeza-Loaeza, J.; Ortega, A. Microglial Activation in Metal Neurotoxicity: Impact in Neurodegenerative Diseases. BioMed Res. Int. 2023, 2023, 1–27. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Kotliarova, A.; Sidorova, Y.A. Glial Cell Line-Derived Neurotrophic Factor Family Ligands, Players at the Interface of Neuroinflammation and Neuroprotection: Focus Onto the Glia. Front. Cell. Neurosci. 2021, 15, 679034. [Google Scholar] [CrossRef] [PubMed]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Freude, K. Astrocytes: The Stars in Neurodegeneration? Biomolecules 2024, 14, 289. [Google Scholar] [CrossRef] [PubMed]
- Heir, R.; Abbasi, Z.; Komal, P.; Altimimi, H.F.; Franquin, M.; Moschou, D.; Chambon, J.; Stellwagen, D. Astrocytes Are the Source of TNF Mediating Homeostatic Synaptic Plasticity. J. Neurosci. 2024, 44, e2278222024. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C. Astrocyte Pathology in Major Depressive Disorder: Insights from Human Postmortem Brain Tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Pamphlett, R.; Bishop, D.P. The Toxic Metal Hypothesis for Neurological Disorders. Front. Neurol. 2023, 14, 1173779. [Google Scholar] [CrossRef]
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro 2020, 12, 175909142096268. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xia, M.; Zorec, R.; Parpura, V.; Verkhratsky, A. Astrocytes in Heavy Metal Neurotoxicity and Neurodegeneration. Brain Res. 2021, 1752, 147234. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Bodnya, C.; Ilieva, I.; Neely, M.D.; Aschner, M.; Bowman, A.B. Huntington’s Disease Associated Resistance to Mn Neurotoxicity Is Neurodevelopmental Stage and Neuronal Lineage Dependent. Neurotoxicology 2019, 75, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Interaction of Heavy Metal Lead with Gut Microbiota: Implications for Autism Spectrum Disorder. Biomolecules 2023, 13, 1549. [Google Scholar] [CrossRef]
- Michalski, J.-P.; Kothary, R. Oligodendrocytes in a Nutshell. Front. Cell. Neurosci. 2015, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Nomden, A.; Van Noort, J.M.; Baron, W. Toll-like Receptors 2 and 3 Agonists Differentially Affect Oligodendrocyte Survival, Differentiation, and Myelin Membrane Formation. J. Neurosci. Res. 2012, 90, 388–398. [Google Scholar] [CrossRef]
- Kumar, V. Toll-Like Receptors in Adaptive Immunity. In Toll-like Receptors in Health and Disease; Kumar, V., Ed.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2021; Volume 276, pp. 95–131. ISBN 978-3-031-06511-8. [Google Scholar]
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like Receptors 2 and 4 Differentially Regulate the Self-Renewal and Differentiation of Spinal Cord Neural Precursor Cells. Stem Cell Res. Ther. 2022, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tong, H.; Yang, T.; Liu, L.; Li, X.-J.; Li, S. Insights into White Matter Defect in Huntington’s Disease. Cells 2022, 11, 3381. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Macrì, R.; Bava, I.; Gliozzi, M.; Musolino, V.; Nucera, S.; Carresi, C.; Scicchitano, M.; Bosco, F.; Scarano, F.; et al. Myelin Disturbances Produced by Sub-Toxic Concentration of Heavy Metals: The Role of Oligodendrocyte Dysfunction. Int. J. Mol. Sci. 2019, 20, 4554. [Google Scholar] [CrossRef]
- Hill, R.A.; Patel, K.D.; Goncalves, C.M.; Grutzendler, J.; Nishiyama, A. Modulation of Oligodendrocyte Generation during a Critical Temporal Window after NG2 Cell Division. Nat. Neurosci. 2014, 17, 1518–1527. [Google Scholar] [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Dimou, L.; Gallo, V. NG 2-glia and Their Functions in the Central Nervous System. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wang, W.; Zhou, M. Spatial Organization of NG2 Glial Cells and Astrocytes in Rat Hippocampal CA1 Region. Hippocampus 2014, 24, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a Common Denominator for Neuroinflammation, Blood–Brain Barrier Alteration, and Oligodendrocyte Precursor Response in EAE, Plays a Role in Dendritic Cell Activation. Acta Neuropathol. 2016, 132, 23–42. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Q.; Yang, Q.; Gu, H.; Yin, Y.; Li, Y.; Hou, J.; Chen, R.; Sun, Q.; Sun, Y.; et al. NG2 Glia Regulate Brain Innate Immunity via TGF-Β2/TGFBR2 Axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef]
- Hu, X.; Geng, P.; Zhao, X.; Wang, Q.; Liu, C.; Guo, C.; Dong, W.; Jin, X. The NG2-Glia Is a Potential Target to Maintain the Integrity of Neurovascular Unit after Acute Ischemic Stroke. Neurobiol. Dis. 2023, 180, 106076. [Google Scholar] [CrossRef]
- Timmermann, A.; Tascio, D.; Jabs, R.; Boehlen, A.; Domingos, C.; Skubal, M.; Huang, W.; Kirchhoff, F.; Henneberger, C.; Bilkei-Gorzo, A.; et al. Dysfunction of NG2 Glial Cells Affects Neuronal Plasticity and Behavior. Glia 2023, 71, 1481–1501. [Google Scholar] [CrossRef]
- Vélez-Fort, M.; Maldonado, P.P.; Butt, A.M.; Audinat, E.; Angulo, M.C. Postnatal Switch from Synaptic to Extrasynaptic Transmission between Interneurons and NG2 Cells. J. Neurosci. 2010, 30, 6921–6929. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, W.R.; Chen, D.; Garud, N.R. Comparative Population Genetics in the Human Gut Microbiome. Genome Biol. Evol. 2022, 14, evab116. [Google Scholar] [CrossRef]
- Chatterjee, G.; Negi, S.; Basu, S.; Faintuch, J.; O’Donovan, A.; Shukla, P. Microbiome Systems Biology Advancements for Natural Well-Being. Sci. Total Environ. 2022, 838, 155915. [Google Scholar] [CrossRef] [PubMed]
- VanEvery, H.; Franzosa, E.A.; Nguyen, L.H.; Huttenhower, C. Microbiome Epidemiology and Association Studies in Human Health. Nat. Rev. Genet. 2023, 24, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut Reactions: How the Blood–Brain Barrier Connects the Microbiome and the Brain. Exp. Biol. Med. 2018, 243, 159–165. [Google Scholar] [CrossRef]
- Hrncir, T. Gut Microbiota Dysbiosis: Triggers, Consequences, Diagnostic and Therapeutic Options. Microorganisms 2022, 10, 578. [Google Scholar] [CrossRef]
- Sharma, G.; Biswas, S.S.; Mishra, J.; Navik, U.; Kandimalla, R.; Reddy, P.H.; Bhatti, G.K.; Bhatti, J.S. Gut Microbiota Dysbiosis and Huntington’s Disease: Exploring the Gut-Brain Axis and Novel Microbiota-Based Interventions. Life Sci. 2023, 328, 121882. [Google Scholar] [CrossRef]
- Tong, H.; Yang, T.; Xu, S.; Li, X.; Liu, L.; Zhou, G.; Yang, S.; Yin, S.; Li, X.-J.; Li, S. Huntington’s Disease: Complex Pathogenesis and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 3845. [Google Scholar] [CrossRef]
- Strandwitz, P. Neurotransmitter Modulation by the Gut Microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.-J.; Zhu, Z.-Q. To Re-Examine the Intersection of Microglial Activation and Neuroinflammation in Neurodegenerative Diseases from the Perspective of Pyroptosis. Front. Aging Neurosci. 2023, 15, 1284214. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.G.; Motta, J.-P.; Allain, T.; Ferraz, J.; Wallace, J.L. Pathobiont Release from Dysbiotic Gut Microbiota Biofilms in Intestinal Inflammatory Diseases: A Role for Iron? J. Biomed. Sci. 2019, 26, 1. [Google Scholar] [CrossRef] [PubMed]
- Follmer, C. Gut Microbiome Imbalance and Neuroinflammation: Impact of COVID-19 on Parkinson’s Disease. Mov. Disord. 2020, 35, 1495–1496. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef]
- Canani, R.B. Potential Beneficial Effects of Butyrate in Intestinal and Extraintestinal Diseases. World J. Gastroenterol. 2011, 17, 1519. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak-Wiercioch, A.; Sałat, K. Lipopolysaccharide-Induced Model of Neuroinflammation: Mechanisms of Action, Research Application and Future Directions for Its Use. Molecules 2022, 27, 5481. [Google Scholar] [CrossRef]
- Mitrea, L.; Nemeş, S.-A.; Szabo, K.; Teleky, B.-E.; Vodnar, D.-C. Guts Imbalance Imbalances the Brain: A Review of Gut Microbiota Association With Neurological and Psychiatric Disorders. Front. Med. 2022, 9, 813204. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Leaky Gut: Mechanisms, Measurement and Clinical Implications in Humans. Gut 2019, 68, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.I.; Mercieca, E.-C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut Dysbiosis in Huntington’s Disease: Associations among Gut Microbiota, Cognitive Performance and Clinical Outcomes. Brain Commun. 2020, 2, fcaa110. [Google Scholar] [CrossRef]
- Witkowska, D.; Słowik, J.; Chilicka, K. Heavy Metals and Human Health: Possible Exposure Pathways and the Competition for Protein Binding Sites. Molecules 2021, 26, 6060. [Google Scholar] [CrossRef]
- Koch, W.; Czop, M.; Iłowiecka, K.; Nawrocka, A.; Wiącek, D. Dietary Intake of Toxic Heavy Metals with Major Groups of Food Products—Results of Analytical Determinations. Nutrients 2022, 14, 1626. [Google Scholar] [CrossRef]
- Rieder, G.S.; Duarte, T.; Delgado, C.P.; Rodighiero, A.; Nogara, P.A.; Orian, L.; Aschner, M.; Dalla Corte, C.L.; Da Rocha, J.B.T. Interplay between Diphenyl Diselenide and Copper: Impact on D. Melanogaster Survival, Behavior, and Biochemical Parameters. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2024, 281, 109899. [Google Scholar] [CrossRef]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Yousefi Babadi, V.; Sadeghi, L.; Shirani, K.; Malekirad, A.A.; Rezaei, M. The Toxic Effect of Manganese on the Acetylcholinesterase Activity in Rat Brains. J. Toxicol. 2014, 2014, 946372. [Google Scholar] [CrossRef]
- Horning, K.J.; Caito, S.W.; Tipps, K.G.; Bowman, A.B.; Aschner, M. Manganese Is Essential for Neuronal Health. Annu. Rev. Nutr. 2015, 35, 71–108. [Google Scholar] [CrossRef]
- Andrade, V.; Mateus, M.L.; Batoréu, M.C.; Aschner, M.; Dos Santos, A.M. Toxic Mechanisms Underlying Motor Activity Changes Induced by a Mixture of Lead, Arsenic and Manganese. EC Pharmacol. Toxicol. 2017, 3, 31–42. [Google Scholar] [PubMed]
- Peres, T.V.; Schettinger, M.R.C.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-Induced Neurotoxicity: A Review of Its Behavioral Consequences and Neuroprotective Strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, S.L.; Zheng, W. Manganese Toxicity Upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef]
- Burton, N.C.; Schneider, J.S.; Syversen, T.; Guilarte, T.R. Effects of Chronic Manganese Exposure on Glutamatergic and GABAergic Neurotransmitter Markers in the Nonhuman Primate Brain. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 111, 131–139. [Google Scholar] [CrossRef]
- Aschner, M.; Gannon, M. Manganese (Mn) Transport across the Rat Blood-Brain Barrier: Saturable and Transferrin-Dependent Transport Mechanisms. Brain Res. Bull. 1994, 33, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zheng, K.; Li, S.; Ren, C.; Shen, Y.; Tian, L.; Zhu, H.; Zhou, Z.; Jiang, Y. Insight into the Potential Role of Ferroptosis in Neurodegenerative Diseases. Front. Cell. Neurosci. 2022, 16, 1005182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]
- Tian, H.-Y.; Huang, B.-Y.; Nie, H.-F.; Chen, X.-Y.; Zhou, Y.; Yang, T.; Cheng, S.-W.; Mei, Z.-G.; Ge, J.-W. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sci. 2023, 13, 1367. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Tang, D.; Wang, Y.; Li, X.; Bao, H.; Tang, C.; Dong, X.; Li, X.; Yang, Q.; Yan, Y.; et al. The Mechanism of Ferroptosis and Its Related Diseases. Mol. Biomed. 2023, 4, 33. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.d.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Johnson, E.B.; Parker, C.S.; Scahill, R.I.; Gregory, S.; Papoutsi, M.; Zeun, P.; Osborne-Crowley, K.; Lowe, J.; Nair, A.; Estevez-Fraga, C.; et al. Altered Iron and Myelin in Premanifest Huntington’s Disease More than 20 Years before Clinical Onset: Evidence from the Cross-Sectional HD Young Adult Study. EBioMedicine 2021, 65, 103266. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Ripamonti, M.; Moro, A.S.; Cozzi, A. Iron Imbalance in Neurodegeneration. Mol. Psychiatry 2024, 29, 1139–1152. [Google Scholar] [CrossRef]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxidants 2021, 10, 1864. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Liu, H.; Shi, X.-J.; Cheng, Y. Blood Oxidative Stress Marker Aberrations in Patients with Huntington’s Disease: A Meta-Analysis Study. Oxid. Med. Cell. Longev. 2020, 2020, 9187195. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, G.; Liang, Z.; Qi, T.; Deng, K.; Yu, J.; Peng, Y.; Zheng, J.; Song, Y.; Chang, X. Microbiota-Assisted Iron Uptake Promotes Immune Tolerance in the Intestine. Nat. Commun. 2023, 14, 2790. [Google Scholar] [CrossRef] [PubMed]
- Correnti, M.; Gammella, E.; Cairo, G.; Recalcati, S. Iron Absorption: Molecular and Pathophysiological Aspects. Metabolites 2024, 14, 228. [Google Scholar] [CrossRef]
- Patanè, G.T.; Putaggio, S.; Tellone, E.; Barreca, D.; Ficarra, S.; Maffei, C.; Calderaro, A.; Laganà, G. Ferroptosis: Emerging Role in Diseases and Potential Implication of Bioactive Compounds. Int. J. Mol. Sci. 2023, 24, 17279. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting Ferroptosis Opens New Avenues for the Development of Novel Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Ji, M.; Wu, C.; Zhang, Y.; Ji, S. Targeting Ferroptosis in Neuroimmune and Neurodegenerative Disorders for the Development of Novel Therapeutics. Biomed. Pharmacother. 2024, 176, 116777. [Google Scholar] [CrossRef] [PubMed]
- Aschner, J.L.; Aschner, M. Nutritional Aspects of Manganese Homeostasis. Mol. Aspects Med. 2005, 26, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxid. Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Getachew, B.; Aschner, M. Butyrate Protects and Synergizes with Nicotine against Iron- and Manganese-Induced Toxicities in Cell Culture. Neurotox. Res. 2024, 42, 3. [Google Scholar] [CrossRef]
- Evans, G.R.; Masullo, L.N. Manganese Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Porru, S.; Esplugues, A.; Llop, S.; Delgado-Saborit, J.M. The Effects of Heavy Metal Exposure on Brain and Gut Microbiota: A Systematic Review of Animal Studies. Environ. Pollut. 2024, 348, 123732. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Martins, A.C.; Oliveira-Paula, G.H.; Skalny, A.V.; Zaitseva, I.P.; Bowman, A.B.; Kirichuk, A.A.; Santamaria, A.; Tizabi, Y.; Tinkov, A.A. Manganese in Autism Spectrum Disorder and Attention Deficit Hyperactivity Disorder: The State of the Art. Curr. Res. Toxicol. 2024, 6, 100170. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.R.; Bowman, A.B. Manganese and the Insulin-IGF Signaling Network in Huntington’s Disease and Other Neurodegenerative Disorders. In Neurotoxicity of Metals; Aschner, M., Costa, L.G., Eds.; Advances in Neurobiology; Springer International Publishing: Cham, Switzerland, 2017; Volume 18, pp. 113–142. ISBN 978-3-319-60188-5. [Google Scholar]
- Cordeiro, L.M.; Soares, M.V.; Da Silva, A.F.; Dos Santos, L.V.; De Souza, L.I.; Da Silveira, T.L.; Baptista, F.B.O.; De Oliveira, G.V.; Pappis, C.; Dressler, V.L.; et al. Toxicity of Copper and Zinc Alone and in Combination in Caenorhabditis Elegans Model of Huntington’s Disease and Protective Effects of Rutin. Neurotoxicology 2023, 97, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Royer, A.; Sharman, T. Copper Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Opazo, C.M.; Lotan, A.; Xiao, Z.; Zhang, B.; Greenough, M.A.; Lim, C.M.; Trytell, H.; Ramírez, A.; Ukuwela, A.A.; Mawal, C.H.; et al. Nutrient Copper Signaling Promotes Protein Turnover by Allosteric Activation of Ubiquitin E2D Conjugases. bioRxiv 2021. [Google Scholar]
- Immergluck, J.; Anilkumar, A.C. Wilson Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Xiao, G.; Fan, Q.; Wang, X.; Zhou, B. Huntington Disease Arises from a Combinatory Toxicity of Polyglutamine and Copper Binding. Proc. Natl. Acad. Sci. USA 2013, 110, 14995–15000. [Google Scholar] [CrossRef]
- Pfalzer, A.C.; Yan, Y.; Kang, H.; Totten, M.; Silverman, J.; Bowman, A.B.; Erikson, K.; Claassen, D.O. Alterations in Metal Homeostasis Occur Prior to Canonical Markers in Huntington Disease. Sci. Rep. 2022, 12, 10373. [Google Scholar] [CrossRef]
- Rosas, H.D.; Chen, Y.I.; Doros, G.; Salat, D.H.; Chen, N.; Kwong, K.K.; Bush, A.; Fox, J.; Hersch, S.M. Alterations in Brain Transition Metals in Huntington Disease: An Evolving and Intricate Story. Arch. Neurol. 2012, 69, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Research 2016, 5, 366. [Google Scholar] [CrossRef] [PubMed]
- Kwakye, G.F.; Jiménez, J.A.; Thomas, M.G.; Kingsley, B.A.; McIIvin, M.; Saito, M.A.; Korley, E.M. Heterozygous Huntingtin Promotes Cadmium Neurotoxicity and Neurodegeneration in Striatal Cells via Altered Metal Transport and Protein Kinase C Delta Dependent Oxidative Stress and Apoptosis Signaling Mechanisms. Neurotoxicology 2019, 70, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Haidar, Z.; Fatema, K.; Shoily, S.S.; Sajib, A.A. Disease-Associated Metabolic Pathways Affected by Heavy Metals and Metalloid. Toxicol. Rep. 2023, 10, 554–570. [Google Scholar] [CrossRef]
- Suganya, K.; Koo, B.-S. Gut-Brain Axis: Role of Gut Microbiota on Neurological Disorders and How Probiotics/Prebiotics Beneficially Modulate Microbial and Immune Pathways to Improve Brain Functions. Int. J. Mol. Sci. 2020, 21, 7551. [Google Scholar] [CrossRef]
- Love, C.J.; Masson, B.A.; Gubert, C.; Hannan, A.J. The Microbiota-Gut-Brain Axis in Huntington’s Disease. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2022; Volume 167, pp. 141–184. ISBN 978-0-323-99176-6. [Google Scholar]
- Wronka, D.; Karlik, A.; Misiorek, J.O.; Przybyl, L. What the Gut Tells the Brain—Is There a Link between Microbiota and Huntington’s Disease? Int. J. Mol. Sci. 2023, 24, 4477. [Google Scholar] [CrossRef] [PubMed]
- Ekwudo, M.N.; Gubert, C.; Hannan, A.J. The Microbiota–Gut–Brain Axis in Huntington’s Disease: Pathogenic Mechanisms and Therapeutic Targets. FEBS J. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Getachew, B.; Csoka, A.B.; Bhatti, A.; Copeland, R.L.; Tizabi, Y. Butyrate Protects Against Salsolinol-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurotox. Res. 2020, 38, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Getachew, B.; Csoka, A.B.; Garden, A.R.; Copeland, R.L.; Tizabi, Y. Sodium Butyrate Protects Against Ethanol-Induced Toxicity in SH-SY5Y Cell Line. Neurotox. Res. 2021, 39, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Gubert, C.; Love, C.J.; Kodikara, S.; Mei Liew, J.J.; Renoir, T.; Lê Cao, K.-A.; Hannan, A.J. Gene-Environment-Gut Interactions in Huntington’s Disease Mice Are Associated with Environmental Modulation of the Gut Microbiome. iScience 2022, 25, 103687. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhu, H.; Feng, Y.; Guo, R.; Wan, D. The Impact of Gut Microbiota Disorders on the Blood–Brain Barrier. Infect. Drug Resist. 2020, 13, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Vauleon, S.; Schutz, K.; Massonnet, B.; Gruben, N.; Manchester, M.; Buehler, A.; Schick, E.; Boak, L.; Hawellek, D.J. Quantifying Mutant Huntingtin Protein in Human Cerebrospinal Fluid to Support the Development of Huntingtin-Lowering Therapies. Sci. Rep. 2023, 13, 5332. [Google Scholar] [CrossRef]
- Juarez, D.; Handal-Silva, A.; Morán-Perales, J.L.; Torres-Cifuentes, D.M.; Flores, G.; Treviño, S.; Moreno-Rodriguez, A.; Guevara, J.; Diaz, A. New Insights into Sodium Phenylbutyrate as a Pharmacotherapeutic Option for Neurological Disorders. Synapse 2024, 78, e22301. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Y.; Li, X.; Yu, J.-T.; Wang, Y.-J. Therapeutics for Neurodegenerative Diseases by Targeting the Gut Microbiome: From Bench to Bedside. Transl. Neurodegener. 2024, 13, 12. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Heavy Metal Interactions with Neuroglia and Gut Microbiota: Implications for Huntington’s Disease. Cells 2024, 13, 1144. https://doi.org/10.3390/cells13131144
Tizabi Y, Bennani S, El Kouhen N, Getachew B, Aschner M. Heavy Metal Interactions with Neuroglia and Gut Microbiota: Implications for Huntington’s Disease. Cells. 2024; 13(13):1144. https://doi.org/10.3390/cells13131144
Chicago/Turabian StyleTizabi, Yousef, Samia Bennani, Nacer El Kouhen, Bruk Getachew, and Michael Aschner. 2024. "Heavy Metal Interactions with Neuroglia and Gut Microbiota: Implications for Huntington’s Disease" Cells 13, no. 13: 1144. https://doi.org/10.3390/cells13131144
APA StyleTizabi, Y., Bennani, S., El Kouhen, N., Getachew, B., & Aschner, M. (2024). Heavy Metal Interactions with Neuroglia and Gut Microbiota: Implications for Huntington’s Disease. Cells, 13(13), 1144. https://doi.org/10.3390/cells13131144