
Selective Termination of Autophagy-Dependent Cancers

Abstract

1. What Is Autophagy Dependence?
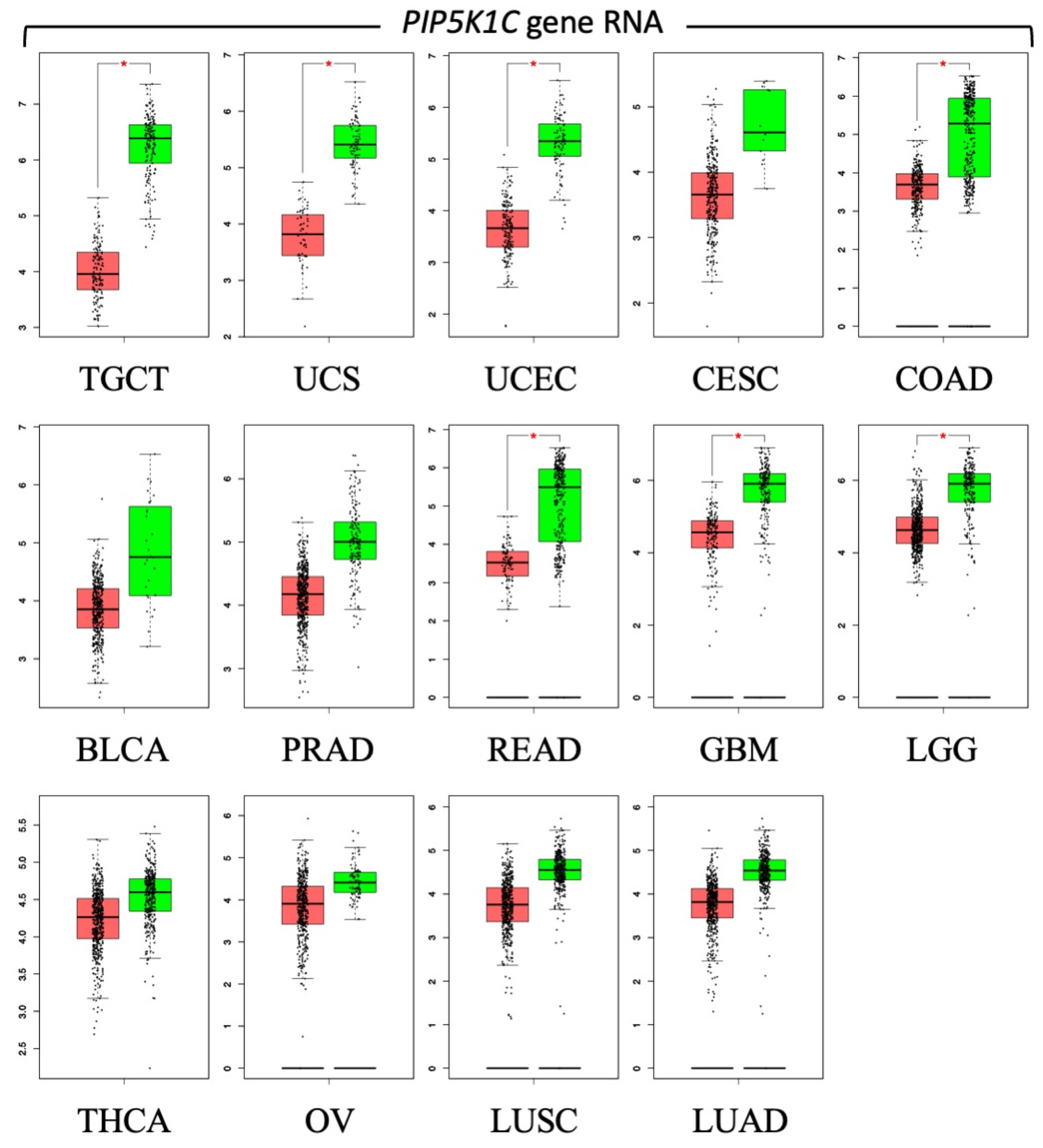
2. What Makes Autophagy-Dependent Cancer Cells PIKFYVE-Dependent?
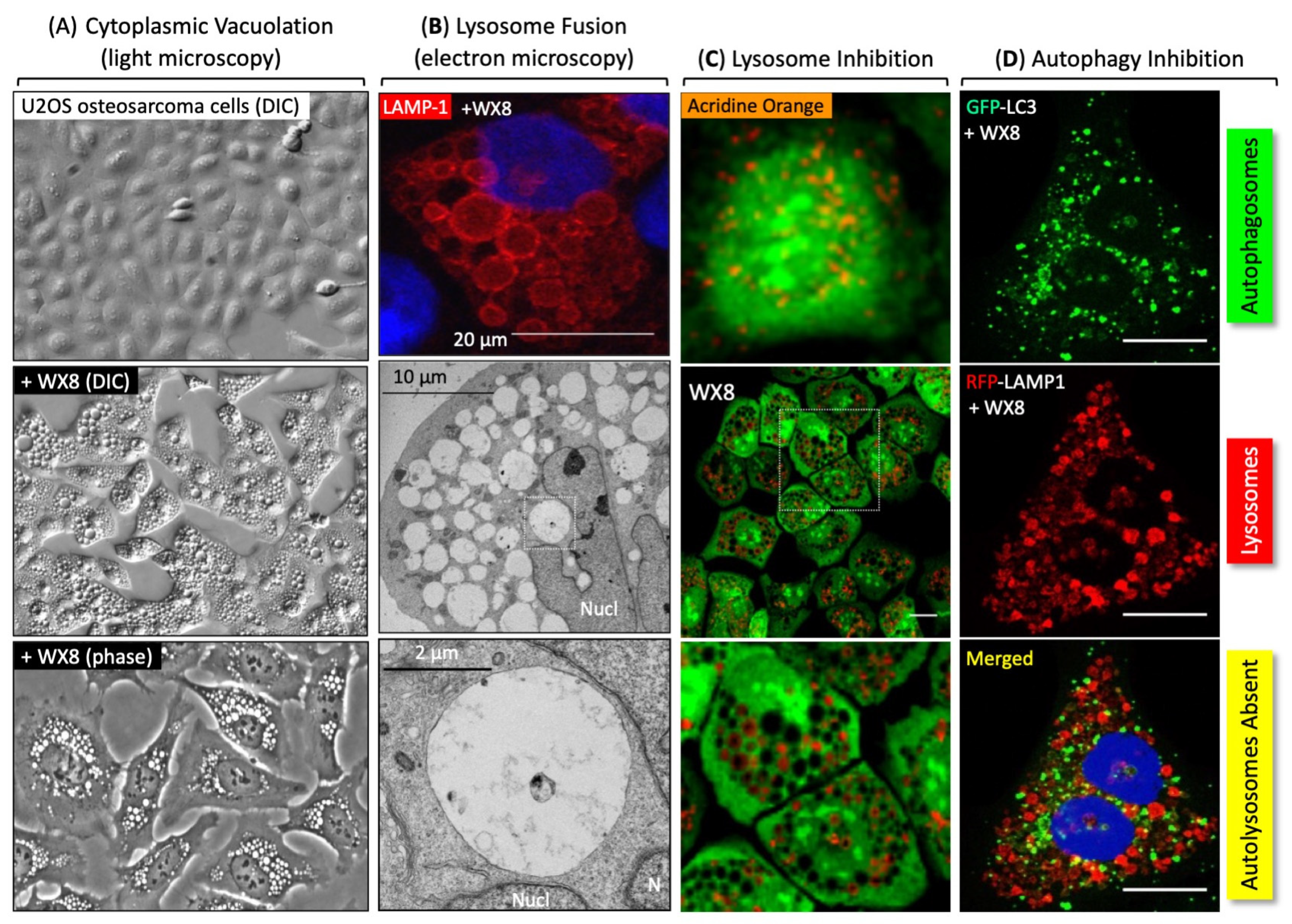
3. The Role of PIKFYVE in Lysosome Homeostasis and Autophagy
4. Current Strategies for Selectively Inhibiting PIKFYVE Activity
- (a)
- Identify small molecules that inhibit PIKFYVE activity
- (b)
- Identify small molecules that inhibit either VAC14 or FIG4
- (c)
- Target the PIKFYVE protein for degradation
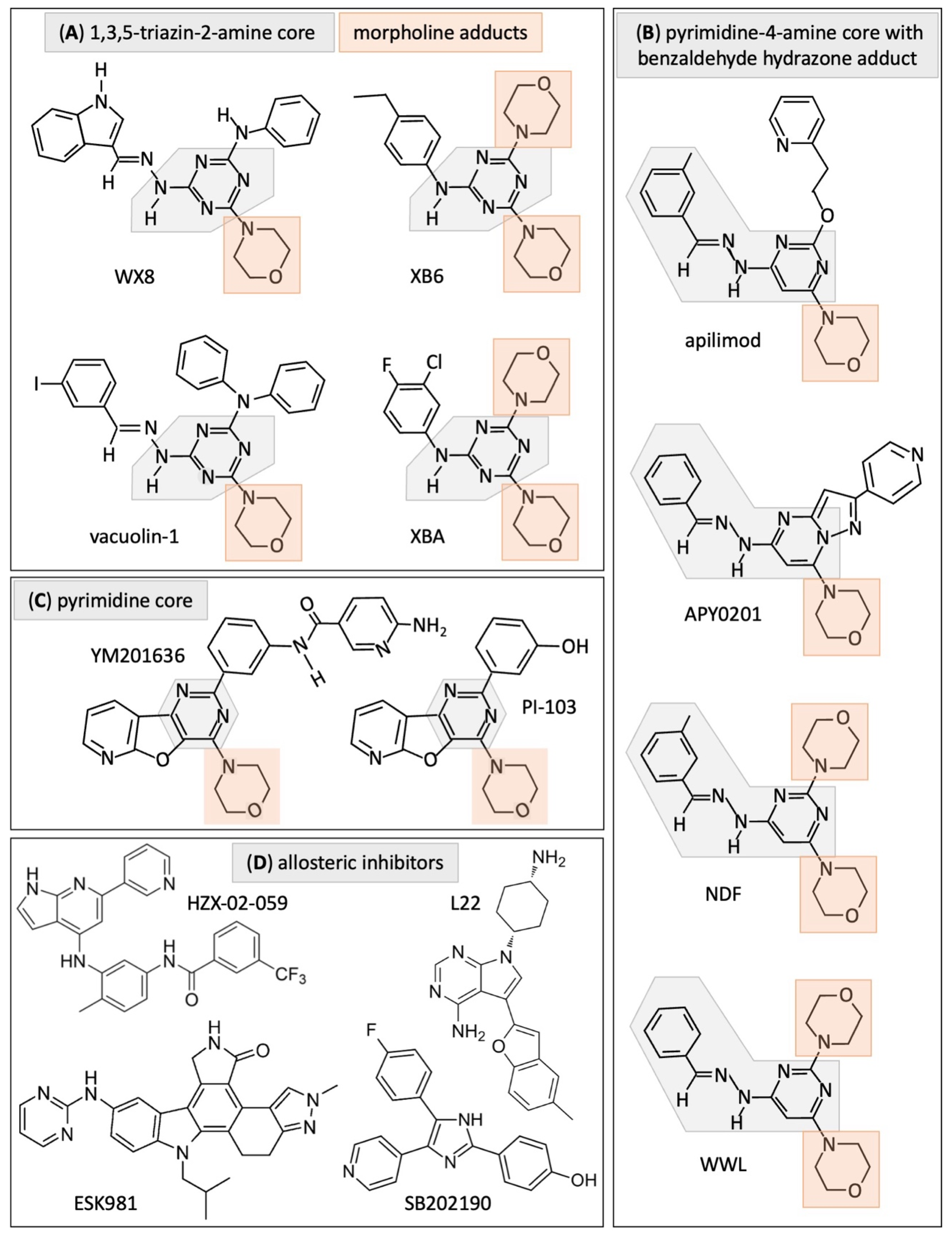
5. PIKFYVE Inhibitors Have Both Primary and Secondary Targets
6. The Significance of Secondary Targets
7. The Effects of PIKFYVE Inhibitors in Immunotherapy
- (a)
- PIKFYVE suppresses the antigen presenting potential of innate immune cells.
- (b)
- PIKFYVE inhibitors facilitate the innate immune response against cancer
8. The Efficacy of PIKFYVE Inhibitors In Vitro
9. The Efficacy of PIKFYVE Inhibitors In Vivo
10. Inhibiting Cell Proliferation Precedes Inducing Cell Death
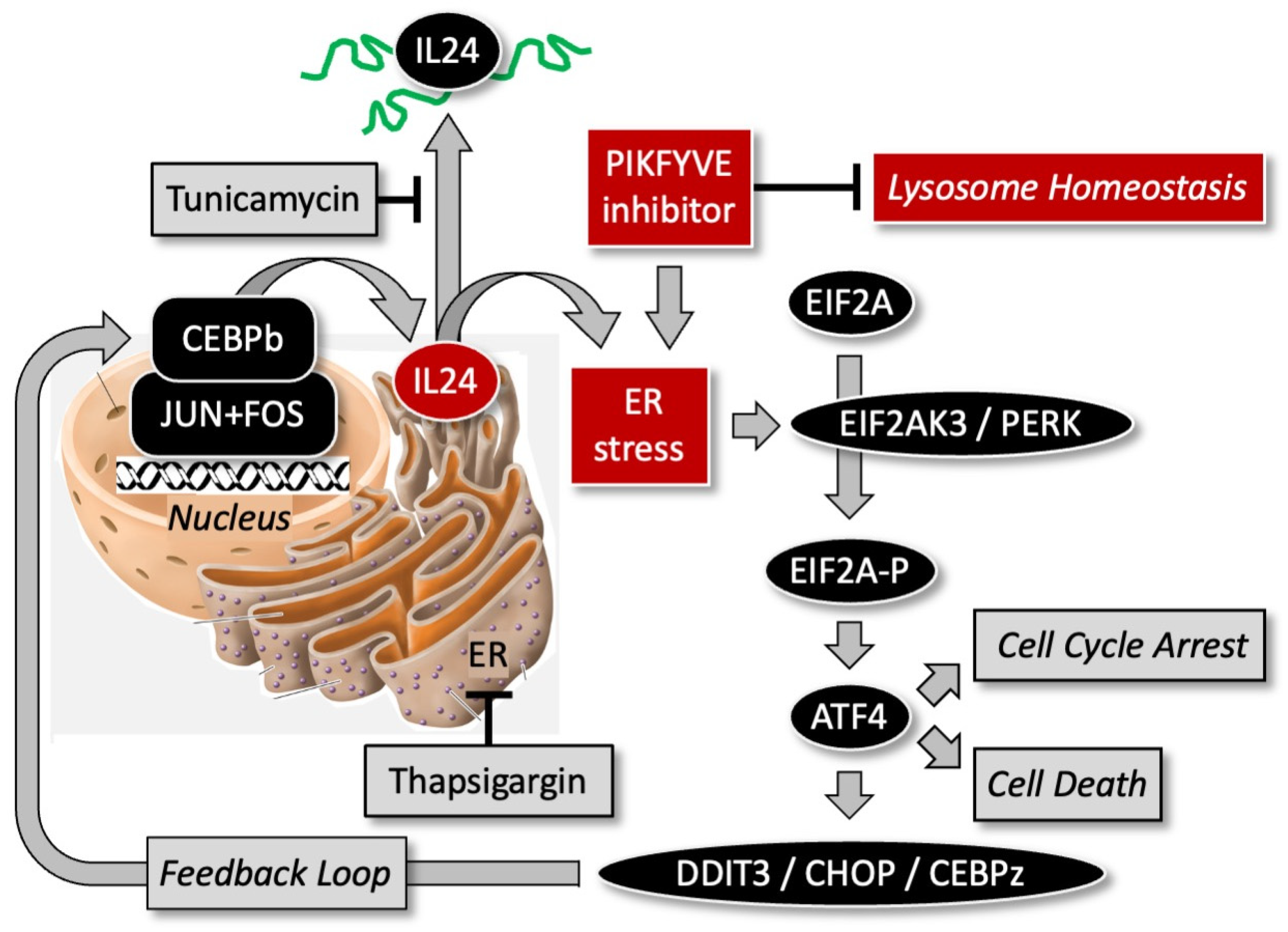
11. PIKFYVE Inhibitors Induce PERK-Dependent Endoplasmic Reticulum Stress
12. PIKFYVE Inhibitors Induce Non-Canonical Apoptosis
13. Therapeutic Potential of PIKFYVE Inhibitors against Cancers
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vassilev, A.; DePamphilis, M.L. Links between DNA Replication, Stem Cells and Cancer. Genes 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Guardia, C.M.; Roy, A.; Vassilev, A.; Saric, A.; Griner, L.N.; Marugan, J.; Ferrer, M.; Bonifacino, J.S.; DePamphilis, M.L. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy 2019, 15, 1694–1718. [Google Scholar] [CrossRef] [PubMed]
- Gayle, S.; Landrette, S.; Beeharry, N.; Conrad, C.; Hernandez, M.; Beckett, P.; Ferguson, S.M.; Mandelkern, T.; Zheng, M.; Xu, T.; et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 2017, 129, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, C.E.; Vassilev, A. Combined Inhibition of p38MAPK and PIKfyve Synergistically Disrupts Autophagy to Selectively Target Cancer Cells. Cancer Res. 2021, 81, 2903–2917. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.R.; Vassilev, A.; Jaiswal, S.K.; O’Connell, C.E.; Ahrens, J.F.; Mallon, B.S.; Pera, M.F.; DePamphilis, M.L. Selective elimination of pluripotent stem cells by PIKfyve specific inhibitors. Stem Cell Rep. 2022, 17, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Choi, J.E.; Tien, J.C.; Simko, S.A.; Rajendiran, T.; Vo, J.N.; Delekta, A.D.; Wang, L.; Xiao, L.; Hodge, N.B.; et al. Autophagy Inhibition by Targeting PIKfyve Potentiates Response to Immune Checkpoint Blockade in Prostate Cancer. Nat. Cancer 2021, 2, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Strohecker, A.M.; White, E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014, 4, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Goodall, M.L.; Wang, T.; Martin, K.R.; Kortus, M.G.; Kauffman, A.L.; Trent, J.M.; Gately, S.; MacKeigan, J.P. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 2014, 10, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Bishop, E.; Bradshaw, T.D. Autophagy modulation: A prudent approach in cancer treatment? Cancer Chemother. Pharmacol. 2018, 82, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyas, E.; Lopez-Bonet, E.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef] [PubMed]
- Nuta, G.C.; Gilad, Y.; Goldberg, N.; Meril, S.; Bahlsen, M.; Carvalho, S.; Kozer, N.; Barr, H.; Fridmann Sirkis, Y.; Hercik, K.; et al. Identifying a selective inhibitor of autophagy that targets ATG12-ATG3 protein-protein interaction. Autophagy 2023, 19, 2372–2385. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Guan, K.L. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- White, E.; Lattime, E.C.; Guo, J.Y. Autophagy Regulates Stress Responses, Metabolism, and Anticancer Immunity. Trends Cancer 2021, 7, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, P.; Strambi, A.; Zipoli, C.; Hagg-Olofsson, M.; Buoncervello, M.; Linder, S.; De Milito, A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: Implications for cancer therapies. Autophagy 2014, 10, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.L.; Pellegrini, P.; Di Lernia, G.; Djavaheri-Mergny, M.; Brnjic, S.; Zhang, X.; Hagg, M.; Linder, S.; Fais, S.; Codogno, P.; et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J. Biol. Chem. 2012, 287, 30664–30676. [Google Scholar] [CrossRef] [PubMed]
- Kuchitsu, Y.; Taguchi, T. Lysosomal microautophagy: An emerging dimension in mammalian autophagy. Trends Cell Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Ikonomov, O.C.; Sbrissa, D.; Shisheva, A. Small molecule PIKfyve inhibitors as cancer therapeutics: Translational promises and limitations. Toxicol. Appl. Pharmacol. 2019, 383, 114771. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, S.; Hao, B.; Li, C.; Zhu, K.; Guo, W.; Wang, Q.; Cheung, K.H.; Wong, C.W.; Wu, W.T.; et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy 2014, 10, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Chakraborty, A.R.; Nomanbhoy, T.; DePamphilis, M.L. PIP5K1C phosphoinositide kinase deficiency distinguishes PIKFYVE-dependent cancer cells from non-malignant cells. Autophagy 2023, 19, 2464–2484. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Chakraborty, A.R.; DePamphilis, M.L. PIKFYVE inhibitors trigger interleukin-24-dependent cell death of autophagy-dependent melanoma. Mol. Oncol. 2024, 18, 988–1011. [Google Scholar] [CrossRef]
- de Campos, C.B.; Zhu, Y.X.; Sepetov, N.; Romanov, S.; Bruins, L.A.; Shi, C.X.; Stein, C.K.; Petit, J.L.; Polito, A.N.; Sharik, M.E.; et al. Identification of PIKfyve kinase as a target in multiple myeloma. Haematologica 2020, 105, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Foth, M.; McMahon, M. Autophagy Inhibition in BRAF-Driven Cancers. Cancers 2021, 13, 3498. [Google Scholar] [CrossRef]
- Kim, M.J.; Woo, S.J.; Yoon, C.H.; Lee, J.S.; An, S.; Choi, Y.H.; Hwang, S.G.; Yoon, G.; Lee, S.J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Nyfeler, B.; Eng, C.H. Revisiting autophagy addiction of tumor cells. Autophagy 2016, 12, 1206–1207. [Google Scholar] [CrossRef] [PubMed]
- Wible, D.J.; Parikh, Z.; Cho, E.J.; Chen, M.D.; Jeter, C.R.; Mukhopadhyay, S.; Dalby, K.N.; Varadarajan, S.; Bratton, S.B. Unexpected inhibition of the lipid kinase PIKfyve reveals an epistatic role for p38 MAPKs in endolysosomal fission and volume control. Cell Death Dis. 2024, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Lenk, G.M.; Meisler, M.H. Altered phenotypes due to genetic interaction between the mouse phosphoinositide biosynthesis genes Fig4 and Pip4k2c. G3 Genes Genomes Genet. 2023, 13, jkad007. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.H.; Saffi, G.; Gray, M.A.; Wallace, C.; Dayam, R.M.; Ou, Z.A.; Lenk, G.; Puertollano, R.; Watkins, S.C.; Botelho, R.J. Lysosome enlargement during inhibition of the lipid kinase PIKfyve proceeds through lysosome coalescence. J. Cell Sci. 2018, 131, jcs213587. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Croise, P.; Heiligenstein, X.; Hurbain, I.; Lenk, G.M.; Kaufman, E.; Sannerud, R.; Annaert, W.; Meisler, M.H.; Weisman, L.S.; et al. The PIKfyve complex regulates the early melanosome homeostasis required for physiological amyloid formation. J. Cell Sci. 2019, 132, jcs229500. [Google Scholar] [CrossRef]
- Baba, T.; Balla, T. Emerging roles of phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate as regulators of multiple steps in autophagy. J. Biochem. 2020, 168, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Houdek, X.; Anderson, R.A. Phosphoinositide 3-kinase pathways and autophagy require phosphatidylinositol phosphate kinases. Adv. Biol. Regul. 2018, 68, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Palamiuc, L.; Ravi, A.; Emerling, B.M. Phosphoinositides in autophagy: Current roles and future insights. FEBS J. 2020, 287, 222–238. [Google Scholar] [CrossRef]
- Barlow-Busch, I.; Shaw, A.L.; Burke, J.E. PI4KA and PIKfyve: Essential phosphoinositide signaling enzymes involved in myriad human diseases. Curr. Opin. Cell Biol. 2023, 83, 102207. [Google Scholar] [CrossRef] [PubMed]
- Maib, H.; Adarska, P.; Hunton, R.; Vines, J.H.; Strutt, D.; Bottanelli, F.; Murray, D.H. Recombinant biosensors for multiplex and super-resolution imaging of phosphoinositides. J. Cell Biol. 2024, 223, 95. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.V.; Burke, J.E. Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr. Opin. Cell Biol. 2020, 63, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Rios, P.; Weisman, L.S. Roles of PIKfyve in multiple cellular pathways. Curr. Opin. Cell Biol. 2022, 76, 102086. [Google Scholar] [CrossRef] [PubMed]
- Zolov, S.N.; Bridges, D.; Zhang, Y.; Lee, W.W.; Riehle, E.; Verma, R.; Lenk, G.M.; Converso-Baran, K.; Weide, T.; Albin, R.L.; et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc. Natl. Acad. Sci. USA 2012, 109, 17472–17477. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagosomes: Biogenesis from scratch? Curr. Opin. Cell Biol. 2005, 17, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Zaurito, A.E.; Abdul-Hamid, S.; Fiume, R.; Faenza, I.; Divecha, N. Phosphatidylinositol 5 Phosphate (PI5P): From Behind the Scenes to the Front (Nuclear) Stage. Int. J. Mol. Sci. 2019, 20, 2080. [Google Scholar] [CrossRef] [PubMed]
- Sano, O.; Kazetani, K.; Funata, M.; Fukuda, Y.; Matsui, J.; Iwata, H. Vacuolin-1 inhibits autophagy by impairing lysosomal maturation via PIKfyve inhibition. FEBS Lett. 2016, 590, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xu, Y.; Cheung, A.K.; Tomlinson, R.C.; Alcazar-Roman, A.; Murphy, L.; Billich, A.; Zhang, B.; Feng, Y.; Klumpp, M.; et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem. Biol. 2013, 20, 912–921. [Google Scholar] [CrossRef]
- Cho, H.; Geno, E.; Patoor, M.; Reid, A.; McDonald, R.; Hild, M.; Jenkins, J.L. Indolyl-Pyridinyl-Propenone-Induced Methuosis through the Inhibition of PIKFYVE. ACS Omega 2018, 3, 6097–6103. [Google Scholar] [CrossRef]
- Hayakawa, N.; Noguchi, M.; Takeshita, S.; Eviryanti, A.; Seki, Y.; Nishio, H.; Yokoyama, R.; Noguchi, M.; Shuto, M.; Shima, Y.; et al. Structure-activity relationship study, target identification, and pharmacological characterization of a small molecular IL-12/23 inhibitor, APY0201. Bioorg Med. Chem. 2014, 22, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jin, X.; Zhang, S.; Li, B.; Zeng, L.; He, Y.; Zhang, C. APY0201 Represses Tumor Growth through Inhibiting Autophagy in Gastric Cancer Cells. J. Oncol. 2022, 2022, 7104592. [Google Scholar] [CrossRef] [PubMed]
- Drewry, D.H.; Potjewyd, F.M.; Bayati, A.; Smith, J.L.; Dickmander, R.J.; Howell, S.; Taft-Benz, S.; Min, S.M.; Hossain, M.A.; Heise, M.; et al. Identification and Utilization of a Chemical Probe to Interrogate the Roles of PIKfyve in the Lifecycle of beta-Coronaviruses. J. Med. Chem. 2022, 65, 12860–12882. [Google Scholar] [CrossRef]
- Jefferies, H.B.; Cooke, F.T.; Jat, P.; Boucheron, C.; Koizumi, T.; Hayakawa, M.; Kaizawa, H.; Ohishi, T.; Workman, P.; Waterfield, M.D.; et al. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008, 9, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, K.; Huang, W.; Jiang, Y.; Sun, X.; Zhou, Y.; Li, L.; Li, Y.; Deng, X.; Xu, B. Pharmacological targeting PIKfyve and tubulin as an effective treatment strategy for double-hit lymphoma. Cell Death Discov. 2022, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, S.; Wei, Y.; Wei, H.; Yuan, X.; Xiong, B.; Tang, M.; Yang, T.; Yang, Z.; Ye, H.; et al. Discovery of Potent and Selective Phosphatidylinositol 3-Phosphate 5-Kinase (PIKfyve) Inhibitors as Methuosis Inducers. J. Med. Chem. 2024, 67, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lee, C.Y.; Johnson, R.L.; Wichterman, J.; Huang, R.; DePamphilis, M.L. An image-based, high-throughput screening assay for molecules that induce excess DNA replication in human cancer cells. Mol. Cancer Res. 2011, 9, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Johnson, R.L.; Wichterman-Kouznetsova, J.; Guha, R.; Ferrer, M.; Tuzmen, P.; Martin, S.E.; Zhu, W.; DePamphilis, M.L. High-throughput screening for genes that prevent excess DNA replication in human cells and for molecules that inhibit them. Methods 2012, 57, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, S.; Hao, B.X.; Li, C.; Zhu, K.Y.; Guo, W.J.; Wang, Q.; Cheung, K.-H.; Wong, C.W.N.; Wu, W.-T.; et al. Erratum. Vacuolin-1 potently and reversibly inhibits autophagy by activating Rab5. Autophagy 2018, 14, 176–177. [Google Scholar] [CrossRef]
- Ye, Z.; Wang, D.; Lu, Y.; He, Y.; Yu, J.; Wei, W.; Chen, C.; Wang, R.; Zhang, L.; Zhang, L.; et al. Vacuolin-1 inhibits endosomal trafficking and metastasis via CapZbeta. Oncogene 2021, 40, 1775–1791. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Zhu, K. Inhibition of glioma cell lysosome exocytosis inhibits glioma invasion. PLoS ONE 2012, 7, e45910. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Lu, R.; Zhou, D.; Chu, J.; Przewloka, T.; Zhang, S.; Li, L.; Wu, Y.; Qin, J.; Balasubramanyam, V.; et al. Selective abrogation of Th1 response by STA-5326, a potent IL-12/IL-23 inhibitor. Blood 2007, 109, 1156–1164. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Kaizawa, H.; Kawaguchi, K.; Ishikawa, N.; Koizumi, T.; Ohishi, T.; Yamano, M.; Okada, M.; Ohta, M.; Tsukamoto, S.; et al. Synthesis and biological evaluation of imidazo [1,2-a]pyridine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med. Chem. 2007, 15, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Kawaguchi, K.; Kaizawa, H.; Koizumi, T.; Ohishi, T.; Yamano, M.; Okada, M.; Ohta, M.; Tsukamoto, S.; Raynaud, F.I.; et al. Synthesis and biological evaluation of sulfonylhydrazone-substituted imidazo[1,2-a]pyridines as novel PI3 kinase p110alpha inhibitors. Bioorg Med. Chem. 2007, 15, 5837–5844. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Guan, X.; Yan, J. Two-pore channel blockade by phosphoinositide kinase inhibitors YM201636 and PI-103 determined by a histidine residue near pore-entrance. Commun. Biol. 2022, 5, 738. [Google Scholar] [CrossRef] [PubMed]
- Hudkins, R.L.; Becknell, N.C.; Zulli, A.L.; Underiner, T.L.; Angeles, T.S.; Aimone, L.D.; Albom, M.S.; Chang, H.; Miknyoczki, S.J.; Hunter, K.; et al. Synthesis and biological profile of the pan-vascular endothelial growth factor receptor/tyrosine kinase with immunoglobulin and epidermal growth factor-like homology domains 2 (VEGF-R/TIE-2) inhibitor 11-(2-methylpropyl)-12,13-dihydro-2-methyl-8-(pyrimidin-2-ylamino)-4H-indazolo[5,4-a]pyrrolo[3,4-c]carbazol-4-one (CEP-11981): A novel oncology therapeutic agent. J. Med. Chem. 2012, 55, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Sun, X.; Li, Y.; He, Z.; Li, L.; Deng, Z.; Huang, X.; Han, S.; Zhang, T.; Zhong, J.; et al. Discovery and Identification of Small Molecules as Methuosis Inducers with in Vivo Antitumor Activities. J. Med. Chem. 2018, 61, 5424–5434. [Google Scholar] [CrossRef]
- Lu, Z.; Lai, Q.; Li, Z.F.; Zhong, M.Y.; Jiang, Y.L.; Feng, L.Y.; Zha, J.; Yao, J.W.; Li, Y.; Deng, X.M.; et al. Novel PIKfyve/Tubulin Dual-target Inhibitor as a Promising Therapeutic Strategy for B-cell Acute Lymphoblastic Leukemia. Curr. Med. Sci. 2024, 44, 298–308. [Google Scholar] [CrossRef]
- Sbrissa, D.; Ikonomov, O.C.; Shisheva, A. PIKfyve lipid kinase is a protein kinase: Downregulation of 5′-phosphoinositide product formation by autophosphorylation. Biochemistry 2000, 39, 15980–15989. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qiao, Y.; Jiang, X.; Liu, L.; Zheng, Y.; Qiu, Y.; Cheng, C.; Zhou, F.; Zhou, Y.; Huang, W.; et al. Discovery of a First-in-Class Degrader for the Lipid Kinase PIKfyve. J. Med. Chem. 2023, 66, 12432–12445. [Google Scholar] [CrossRef] [PubMed]
- Al-Ramahi, I.; Giridharan, S.S.P.; Chen, Y.C.; Patnaik, S.; Safren, N.; Hasegawa, J.; de Haro, M.; Wagner Gee, A.K.; Titus, S.A.; Jeong, H.; et al. Inhibition of PIP4Kgamma ameliorates the pathological effects of mutant huntingtin protein. eLife 2017, 6, e29123. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.C.; Ferreira, A.; Marien, J.; Delay, C.; Lee, E.; Trojanowski, J.Q.; Moechars, D.; Annaert, W.; De Muynck, L. PIKfyve activity is required for lysosomal trafficking of tau aggregates and tau seeding. J. Biol. Chem. 2021, 296, 100636. [Google Scholar] [CrossRef] [PubMed]
- Sbrissa, D.; Ikonomov, O.C.; Filios, C.; Delvecchio, K.; Shisheva, A. Functional dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P2 by means of the PIKfyve inhibitor YM201636. Am. J. Physiol. Cell Physiol. 2012, 303, C436–C446. [Google Scholar] [CrossRef] [PubMed]
- Ikonomov, O.C.; Sbrissa, D.; Shisheva, A. YM201636, an inhibitor of retroviral budding and PIKfyve-catalyzed PtdIns(3,5)P2 synthesis, halts glucose entry by insulin in adipocytes. Biochem. Biophys. Res. Commun. 2009, 382, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhu, C.; Zhang, W.; Ta, N.; Zhang, R.; Liu, L.; Feng, D.; Cheng, H.; Liu, J.; Chen, Q. Mitochondrial PIP3-binding protein FUNDC2 supports platelet survival via AKT signaling pathway. Cell Death Differ. 2019, 26, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.I.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007, 67, 5840–5850. [Google Scholar] [CrossRef] [PubMed]
- Comes, F.; Matrone, A.; Lastella, P.; Nico, B.; Susca, F.C.; Bagnulo, R.; Ingravallo, G.; Modica, S.; Lo Sasso, G.; Moschetta, A.; et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007, 14, 693–702. [Google Scholar] [CrossRef]
- Hasegawa, J.; Tokuda, E.; Yao, Y.; Sasaki, T.; Inoki, K.; Weisman, L.S. PP2A-dependent TFEB activation is blocked by PIKfyve-induced mTORC1 activity. Mol. Biol. Cell 2022, 33, ar26. [Google Scholar] [CrossRef]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [PubMed]
- Blitsman, Y.; Hollander, E.; Benafsha, C.; Yegodayev, K.M.; Hadad, U.; Goldbart, R.; Traitel, T.; Rudich, A.; Elkabets, M.; Kost, J. The Potential of PIP3 in Enhancing Wound Healing. Int. J. Mol. Sci. 2024, 25, 1780. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Reschke, R.; Gajewski, T.F. CXCL9 and CXCL10 bring the heat to tumors. Sci. Immunol. 2022, 7, eabq6509. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Qiao, Y.; Choi, J.E.; Zhang, Y.; Mannan, R.; Cheng, C.; He, T.; Zheng, Y.; Yu, J.; Gondal, M.; et al. Targeting the lipid kinase PIKfyve upregulates surface expression of MHC class I to augment cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2023, 120, e2314416120. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Ito, K.; Miyata, H.; Akira, S.; Kawai, T. Deletion of PIKfyve alters alveolar macrophage populations and exacerbates allergic inflammation in mice. EMBO J. 2017, 36, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Baranov, M.V.; Bianchi, F.; Schirmacher, A.; van Aart, M.A.C.; Maassen, S.; Muntjewerff, E.M.; Dingjan, I.; Ter Beest, M.; Verdoes, M.; Keyser, S.G.L.; et al. The Phosphoinositide Kinase PIKfyve Promotes Cathepsin-S-Mediated Major Histocompatibility Complex Class II Antigen Presentation. iScience 2019, 11, 160–177. [Google Scholar] [CrossRef]
- Cai, X.; Xu, Y.; Kim, Y.M.; Loureiro, J.; Huang, Q. PIKfyve, a class III lipid kinase, is required for TLR-induced type I IFN production via modulation of ATF3. J. Immunol. 2014, 192, 3383–3389. [Google Scholar] [CrossRef] [PubMed]
- Dayam, R.M.; Sun, C.X.; Choy, C.H.; Mancuso, G.; Glogauer, M.; Botelho, R.J. The Lipid Kinase PIKfyve Coordinates the Neutrophil Immune Response through the Activation of the Rac GTPase. J. Immunol. 2017, 199, 2096–2105. [Google Scholar] [CrossRef]
- Choi, J.E.; Qiao, Y.; Kryczek, I.; Yu, J.; Gurkan, J.; Bao, Y.; Gondal, M.; Tien, J.C.; Maj, T.; Yazdani, S.; et al. PIKfyve controls dendritic cell function and tumor immunity. bioRxiv 2024. [Google Scholar] [CrossRef]
- Logue, J.; Chakraborty, A.R.; Johnson, R.; Goyal, G.; Rodas, M.; Taylor, L.J.; Baracco, L.; McGrath, M.E.; Haupt, R.; Furlong, B.A.; et al. PIKfyve-specific inhibitors restrict replication of multiple coronaviruses in vitro but not in a murine model of COVID-19. Commun. Biol. 2022, 5, 808. [Google Scholar] [CrossRef] [PubMed]
- Baranov, M.V.; Bianchi, F.; van den Bogaart, G. The PIKfyve Inhibitor Apilimod: A Double-Edged Sword against COVID-19. Cells 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Z.; Xi, Z.Q.; Niu, J.; Li, W.; Wang, X.; Liang, C.; Sun, H.; Fang, D.; Xie, S.Q. Inhibition of PIKfyve using YM201636 suppresses the growth of liver cancer via the induction of autophagy. Oncol. Rep. 2019, 41, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Ikonomov, O.C.; Sbrissa, D.; Delvecchio, K.; Xie, Y.; Jin, J.P.; Rappolee, D.; Shisheva, A. The phosphoinositide kinase PIKfyve is vital in early embryonic development: Preimplantation lethality of PIKfyve-/- embryos but normality of PIKfyve+/- mice. J. Biol. Chem. 2011, 286, 13404–13413. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; DeWald, D.B.; Boronenkov, I.V.; Anderson, R.A.; Emr, S.D.; Koshland, D. Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol. Biol. Cell 1995, 6, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Sbrissa, D.; Naisan, G.; Ikonomov, O.C.; Shisheva, A. Apilimod, a candidate anticancer therapeutic, arrests not only PtdIns(3,5)P2 but also PtdIns5P synthesis by PIKfyve and induces bafilomycin A1-reversible aberrant endomembrane dilation. PLoS ONE 2018, 13, e0204532. [Google Scholar] [CrossRef] [PubMed]
- Ikonomov, O.C.; Altankov, G.; Sbrissa, D.; Shisheva, A. PIKfyve inhibitor cytotoxicity requires AKT suppression and excessive cytoplasmic vacuolation. Toxicol. Appl. Pharmacol. 2018, 356, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Shacham, T.; Patel, C.; Lederkremer, G.Z. PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 2021, 11, 354. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Dash, R.; Richards, J.E.; Su, Z.Z.; Bhutia, S.K.; Azab, B.; Rahmani, M.; Dasmahapatra, G.; Yacoub, A.; Dent, P.; Dmitriev, I.P.; et al. Mechanism by which Mcl-1 regulates cancer-specific apoptosis triggered by mda-7/IL-24, an IL-10-related cytokine. Cancer Res. 2010, 70, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Yacoub, A.; Sarkar, D.; Emdad, L.; Rahmani, M.; Spiegel, S.; Koumenis, C.; Graf, M.; Curiel, D.T.; Grant, S.; et al. PERK-dependent regulation of MDA-7/IL-24-induced autophagy in primary human glioma cells. Autophagy 2008, 4, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Persaud, L.; Mighty, J.; Zhong, X.; Francis, A.; Mendez, M.; Muharam, H.; Redenti, S.M.; Das, D.; Aktas, B.H.; Sauane, M. IL-24 Promotes Apoptosis through cAMP-Dependent PKA Pathways in Human Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3561. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Mayo, M.; Dash, R.; Sokhi, U.K.; Dmitriev, I.P.; Sarkar, D.; Dent, P.; Curiel, D.T.; Fisher, P.B.; Grant, S. Melanoma differentiation associated gene-7/interleukin-24 potently induces apoptosis in human myeloid leukemia cells through a process regulated by endoplasmic reticulum stress. Mol. Pharmacol. 2010, 78, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Sauane, M.; Su, Z.Z.; Gupta, P.; Lebedeva, I.V.; Dent, P.; Sarkar, D.; Fisher, P.B. Autocrine regulation of mda-7/IL-24 mediates cancer-specific apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9763–9768. [Google Scholar] [CrossRef] [PubMed]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Moller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal 2020, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Luhr, M.; Torgersen, M.L.; Szalai, P.; Hashim, A.; Brech, A.; Staerk, J.; Engedal, N. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J. Biol. Chem. 2019, 294, 8197–8217. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.; Jaiswal, S.K.; DePamphilis, M.L. Cell Death and the p53 Enigma During Mammalian Embryonic Development. Stem Cells 2022, 40, 227–238. [Google Scholar] [CrossRef]
- Martin, S.; Harper, C.B.; May, L.M.; Coulson, E.J.; Meunier, F.A.; Osborne, S.L. Inhibition of PIKfyve by YM-201636 dysregulates autophagy and leads to apoptosis-independent neuronal cell death. PLoS ONE 2013, 8, e60152. [Google Scholar] [CrossRef]
- Liu, X.D.; Yao, J.; Tripathi, D.N.; Ding, Z.; Xu, Y.; Sun, M.; Zhang, J.; Bai, S.; German, P.; Hoang, A.; et al. Autophagy mediates HIF2alpha degradation and suppresses renal tumorigenesis. Oncogene 2015, 34, 2450–2460. [Google Scholar] [CrossRef]
- Huang, X.; Liang, H.; Zhang, H.; Tian, L.; Cong, P.; Wu, T.; Zhang, Q.; Gao, X.; Li, W.; Chen, A.; et al. The Potential Mechanism of Cancer Patients Appearing More Vulnerable to SARS-CoV-2 and Poor Outcomes: A Pan-Cancer Bioinformatics Analysis. Front. Immunol. 2022, 10, 804387. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.T.; Linares, G.R.; Chang, W.H.; Eoh, Y.; Krishnan, G.; Mendonca, S.; Hong, S.; Shi, Y.; Santana, M.; Kueth, C.; et al. PIKFYVE inhibition mitigates disease in models of diverse forms of ALS. Cell 2023, 186, 786–802.e728. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Triscott, J.; Emerling, B.M.; Hammond, G.R.V. Beyond PI3Ks: Targeting phosphoinositide kinases in disease. Nat. Rev. Drug Discov. 2023, 22, 357–386. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef] [PubMed]
- Milan, E.; Fabbri, M.; Cenci, S. Autophagy in Plasma Cell Ontogeny and Malignancy. J. Clin. Immunol. 2016, 36 (Suppl. S1), 18–24. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.K.; Williams, S.A.; Bindra, G.K.; Lay, F.T.; Poon, I.K.H.; Hulett, M.D. Phosphoinositides: Multipurpose cellular lipids with emerging roles in cell death. Cell Death Differ. 2019, 26, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Maruntelu, I.; Constantinescu, A.E.; Covache-Busuioc, R.A.; Constantinescu, I. The Golgi Apparatus: A Key Player in Innate Immunity. Int. J. Mol. Sci. 2024, 25, 4120. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Mukai, K. Innate immunity signalling and membrane trafficking. Curr. Opin. Cell Biol. 2019, 59, 1–7. [Google Scholar] [CrossRef]
- Terajima, M.; Kaneko-Kobayashi, Y.; Nakamura, N.; Yuri, M.; Hiramoto, M.; Naitou, M.; Hattori, K.; Yokota, H.; Mizuhara, H.; Higashi, Y. Inhibition of c-Rel DNA binding is critical for the anti-inflammatory effects of novel PIKfyve inhibitor. Eur. J. Pharmacol. 2016, 780, 93–105. [Google Scholar] [CrossRef]
- Wu, A.; Shi, K.; Wang, J.; Zhang, R.; Wang, Y. Targeting SARS-CoV-2 entry processes: The promising potential and future of host-targeted small-molecule inhibitors. Eur. J. Med. Chem. 2024, 263, 115923. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.; Nicholson, K.A.; Rothstein, J.D.; Swenson, A.; Sampognaro, P.J.; Pant, P.; Macklin, E.A.; Spruill, S.; Paganoni, S.; Gendron, T.F.; et al. Apilimod dimesylate in C9orf72 amyotrophic lateral sclerosis: A randomized phase 2a clinical trial. Brain 2024. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, K.; Kashem, M.; Bauer, A.; Bernardino, A.; Brennan, D.; Cook, B.; Farrow, N.; Molinaro, T.; Nelson, R. Development of Three Orthogonal Assays Suitable for the Identification and Qualification of PIKfyve Inhibitors. Assay. Drug Dev. Technol. 2017, 15, 210–219. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Inhibitor | Selection | Targets | |
|---|---|---|---|---|
| Primary | Secondary | |||
| Kd (nM) | ||||
| A | WX8 | induce excess DNA replication | PIKFYVE 0.9 | PIP4K2C 340 |
| XB6 | PIKFYVE 11 | PIP4K2C 990 | ||
| XBA | PIKFYVE 16 | PIP4K2C 20,000 | ||
| Vacuolin-1 | increase LC3 protein | PIKFYVE 39 | ||
| B | apilimod | reduce cell proliferation | PIKFYVE 0.075, 5.3, 65 | VAC14 |
| APY0201 | inhibit PIK3CA | PIKFYVE | PIK3CA PIK3CB PIK3CD | |
| NDF | excess DNA replication | PIKFYVE 1.6 | PIP4K2C 24,000 | |
| WWL | PIKFYVE 4.8 | PIP4K2C 9200 | ||
| C | YM201636 | inhibit PIK3CA | PIKFYVE 9 | PIK3CA PIK3CB PIK3CD |
| PI-103 | ||||
| D | ESK981 | inhibit receptor tyrosine kinases | PIKFYVE 12 | PIP5K1A 230 PIP5K1C 210 |
| HZX-02-059 | cytoplasmic vacuolization & cell death | PIKFYVE 10 * | PIP4K2C | |
| L22 | PIKFYVE 0.47 | |||
| SB202190 SB203580 | inhibit p38MAPK | p38MAPK | PIKFYVE | |
| Inhibitor | Median IC50 ( µM) | Reference | |||
|---|---|---|---|---|---|
| Cancer Cells | Normal Cells | ||||
| WX8 | 7 different cancers | 0.34 | 4 different | 21 | [27] |
| 10 melanomas | 2.8 | ||||
| 12 different cancers | 0.24 | 8 different | 2.8 | [7] | |
| apilimod | 48 lymphomas | 0.13 | 12 different | 15 | [6] |
| 11 Burkitt’s lymphomas | 0.15 | ||||
| APY0201 | 5 gastric cancers | 0.1–1 | [52] | ||
| YM201636 | 2 hepatocellular carcinomas | 1–5 | [93] | ||
| ESK981 | 7 prostates | 0.08 | [9] | ||
| SB202190 | 12 different cancers | 4.4 | 8 different | 23 | [7] |
| recombinant PIKFYVE protein | 0.4 * | [34] | |||
| 1 prostate | 12.2 ** | ||||
| HZX-02-059 | 3 lymphomas | 0.19 | [55] | ||
| L22 | 1 breast | 0.23 | [56] | ||
| Inhibitor | Cancer | Tumor | Reference |
|---|---|---|---|
| WX8 | melanoma | xenografts | [27] |
| embryonal carcinoma | ex vivo xenografts | [8] | |
| WX8 ± SB202190 | colon adenocarcinoma | xenografts | [7] |
| vacuolin-1 | melanoma breast | transgenic mouse models | [60] |
| apilimod | lymphoma | xenografts | [6] |
| APY0201 | gastric | xenografts | [52] |
| APY0201 apilimod YM201636 | multiple myeloma | ex vivo | [29] |
| YM201636 | hepatocellular carcinoma | xenografts | [93] |
| ESK981 | prostate | ex vivo xenografts | [9] |
| HZX-02-059 | lymphoma | xenografts | [55] |
| L22 | breast | xenografts | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, A.; DePamphilis, M.L. Selective Termination of Autophagy-Dependent Cancers. Cells 2024, 13, 1096. https://doi.org/10.3390/cells13131096
Roy A, DePamphilis ML. Selective Termination of Autophagy-Dependent Cancers. Cells. 2024; 13(13):1096. https://doi.org/10.3390/cells13131096
Chicago/Turabian StyleRoy, Ajit, and Melvin L. DePamphilis. 2024. "Selective Termination of Autophagy-Dependent Cancers" Cells 13, no. 13: 1096. https://doi.org/10.3390/cells13131096
APA StyleRoy, A., & DePamphilis, M. L. (2024). Selective Termination of Autophagy-Dependent Cancers. Cells, 13(13), 1096. https://doi.org/10.3390/cells13131096