
iPSC-Derived Astrocytes and Neurons Replicate Brain Gene Expression, Epigenetic, Cell Morphology and Connectivity Alterations Found in Autism
 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. iPSC Maintenance and Cell Expansion
2.3. Differentiation of iPSCs to Neuronal Stem Cells (NSC), Astrocytes and Neurons
2.4. Immunocytochemistry
2.5. DNA and RNA Extraction and cDNA Synthesis
2.6. Gene Expression Analysis
2.7. DNA Methylation Analysis
2.8. Evaluation of Growth Rate in NSCs and Differentiated Cells and Neuronal Migration Assay
2.9. Statistical Analysis
3. Results
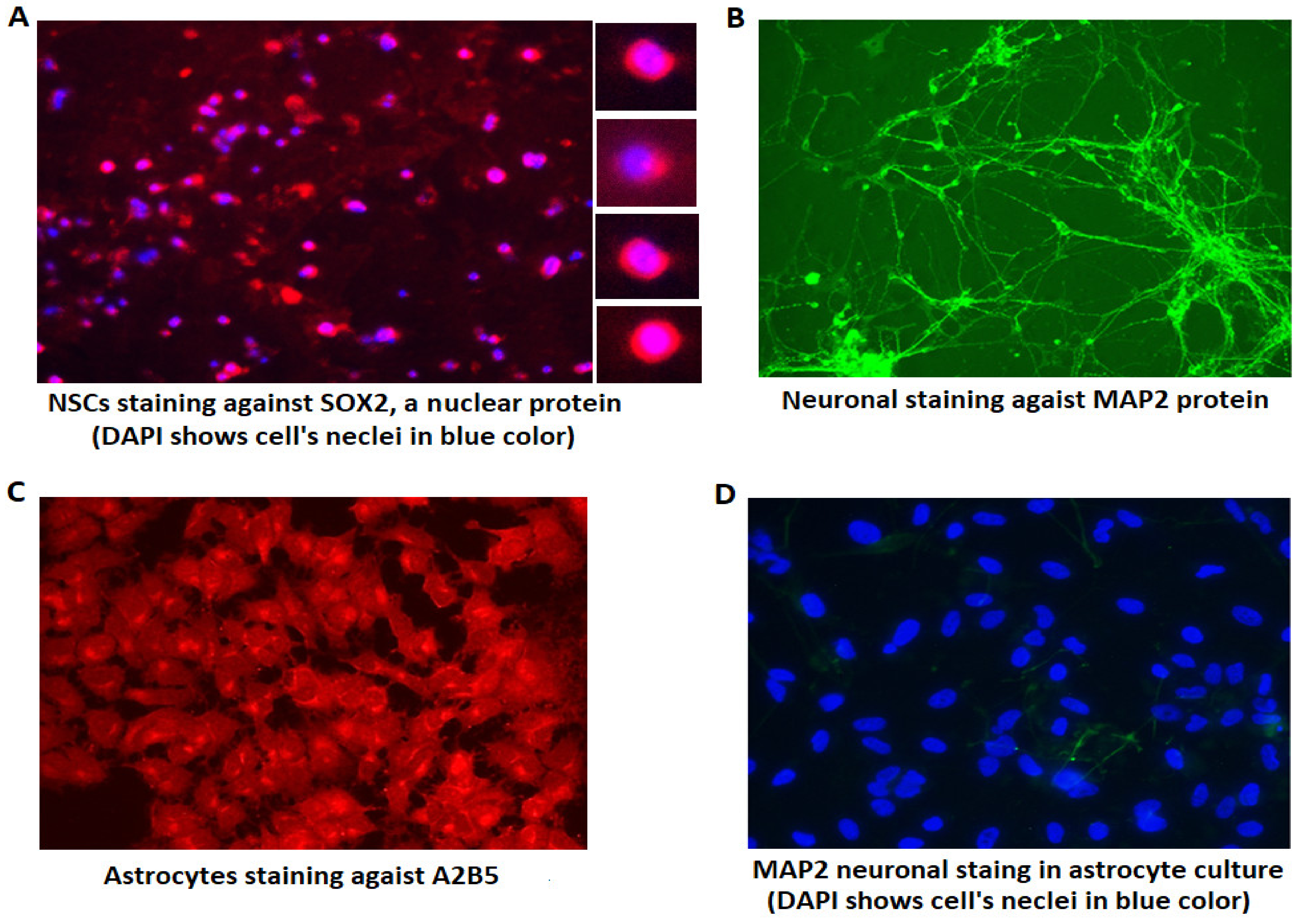
3.1. Verifying the Identity of Differentiated Cells Using Immunohistochemistry
3.2. Expression Alterations of NSCs and Differentiated Cells
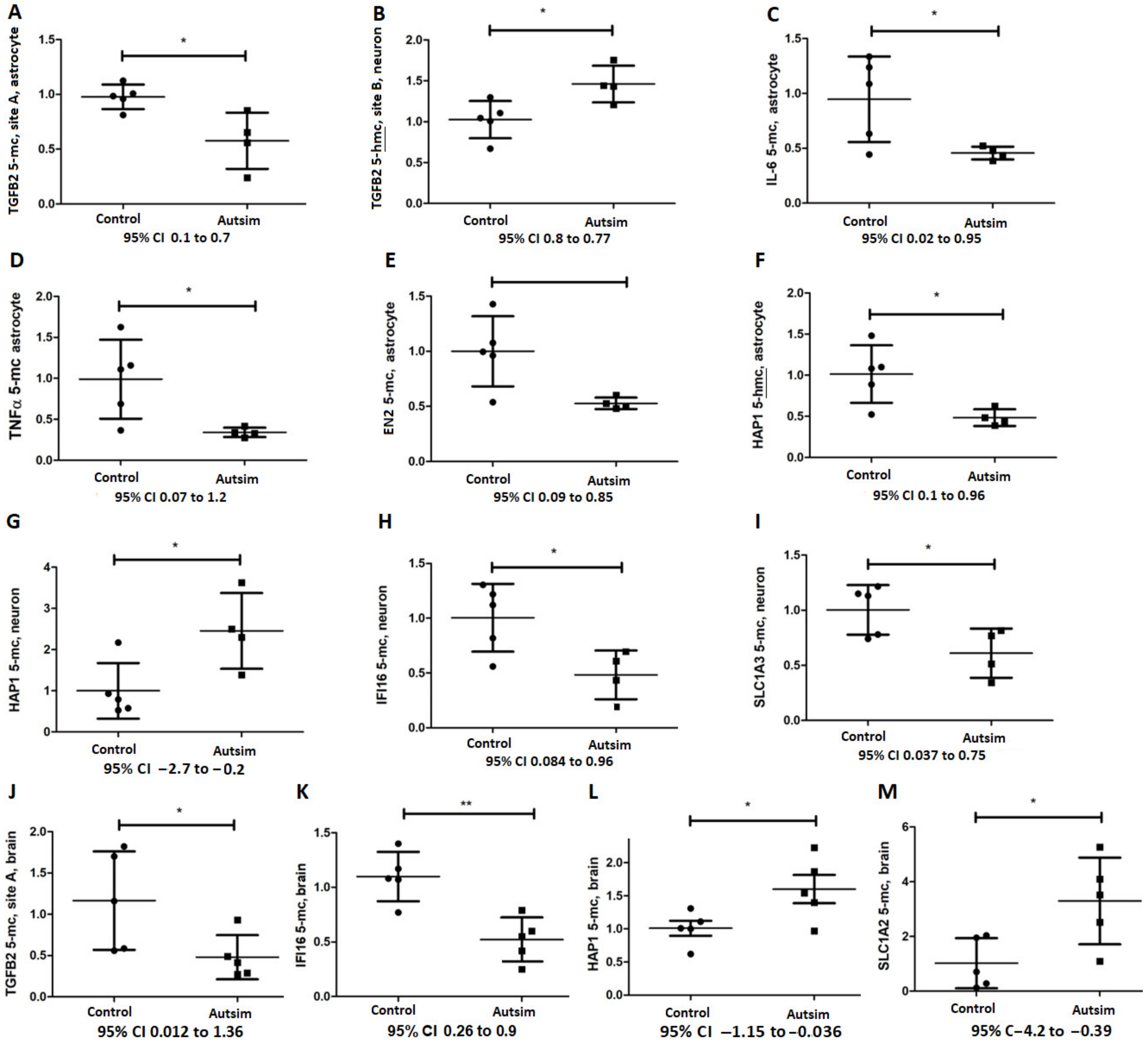
3.3. Epigenetic Analysis of Affected Genes Regulated by DNA Methylation
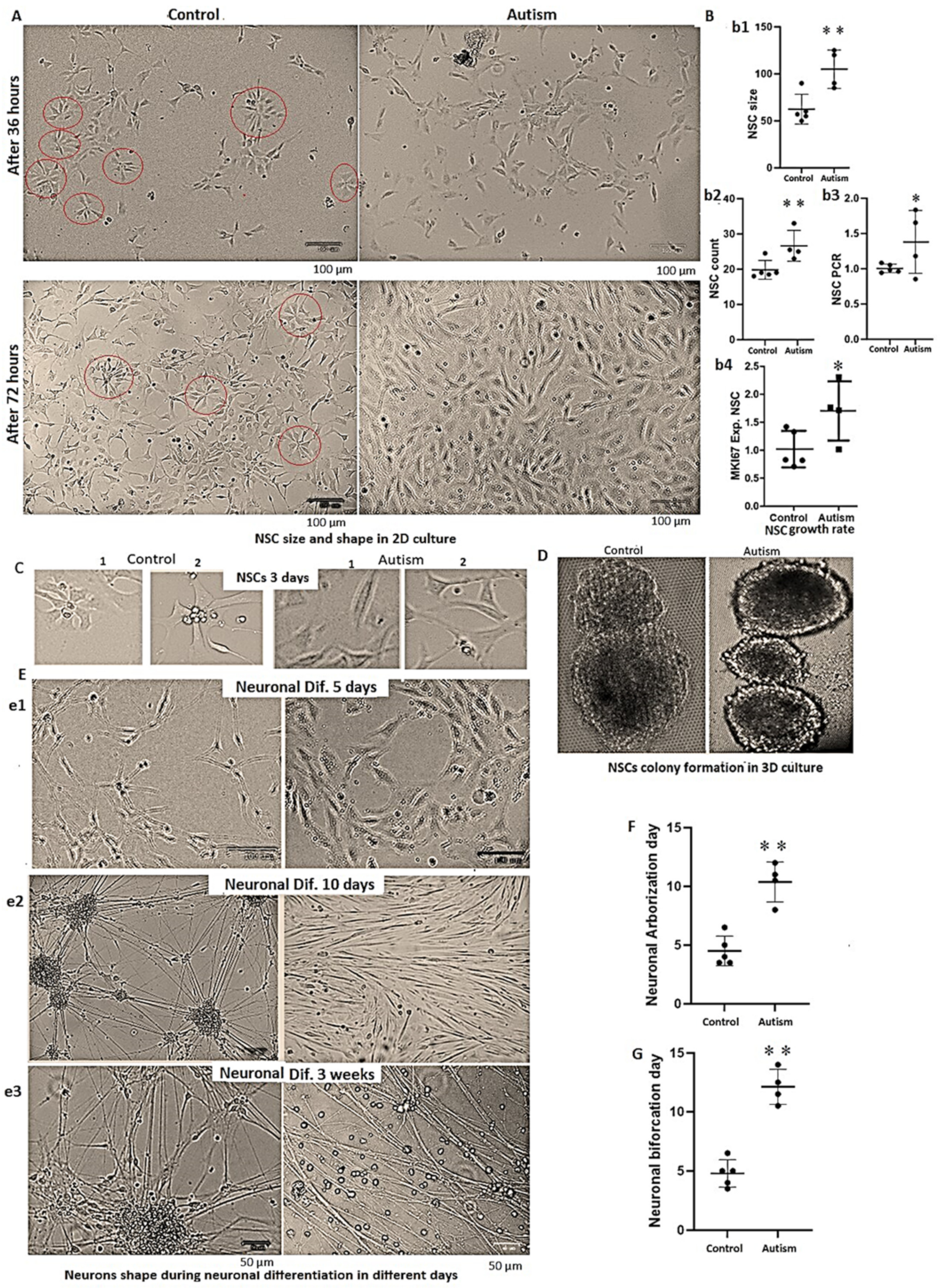
3.4. Alterations in Cells’ Growth Rate and Shape in Autism
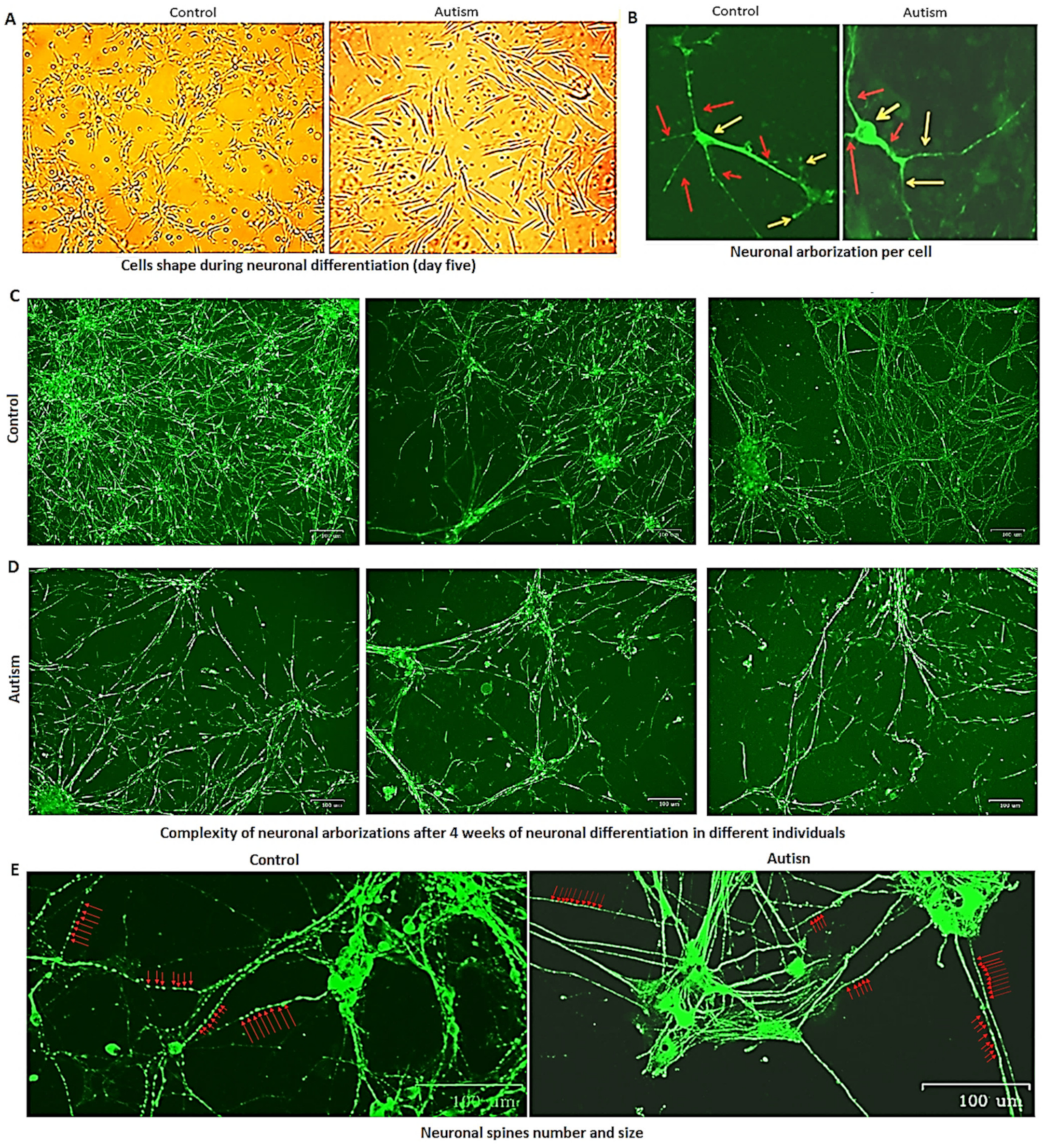
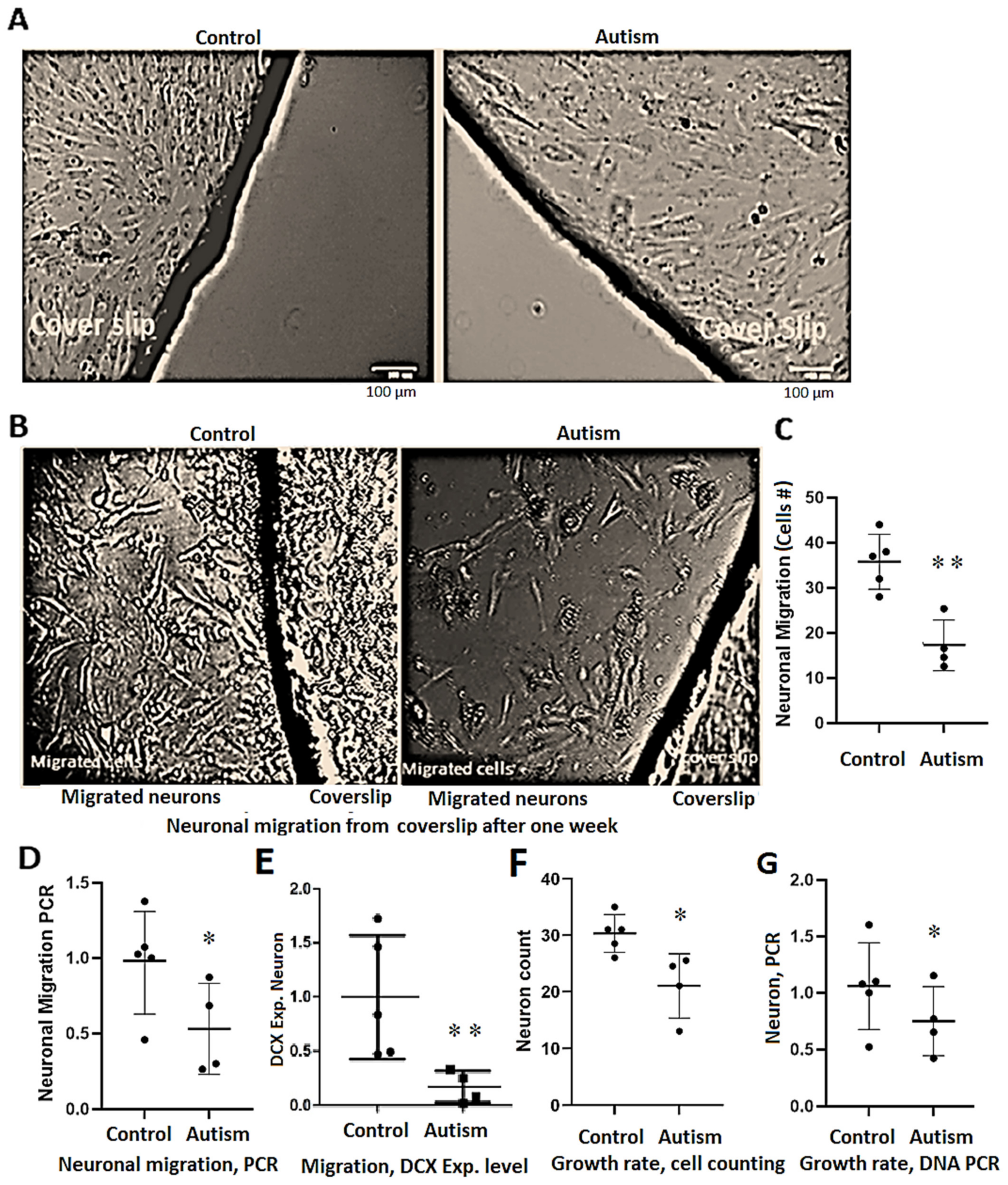
3.5. Delay in Neuronal Differentiation and Reduced Neuronal Migration in Patients with Autism
3.6. Reduced Growth Rate and Long-Term Neuronal Survival in Autism vs. Controls
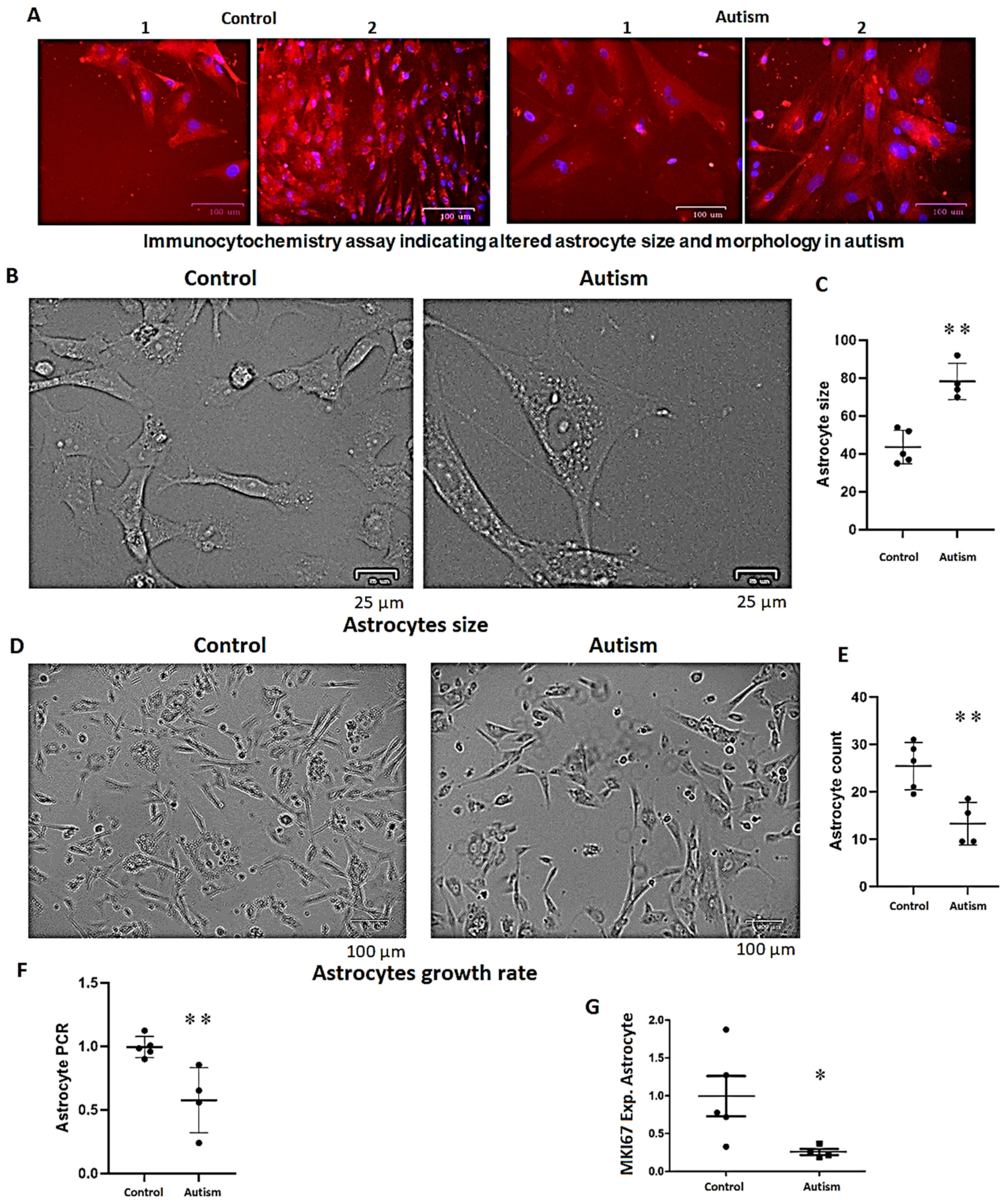
3.7. Increased Astrocyte Size, Reduced Growth Rate and Long-Term Survival in Patients with Autism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 46. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Poe, M.; Gerig, G.; Smith, R.G.; Provenzale, J.; Ross, A.; Gilmore, J.; Piven, J. Magnetic resonance imaging and head circumference study of brain size in autism: Birth through age 2 years. Arch. Gen. Psychiatry 2005, 62, 1366–1376. [Google Scholar] [CrossRef]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef]
- Hardan, A.Y.; Minshew, N.J.; Mallikarjuhn, M.; Keshavan, M.S. Brain volume in autism. J. Child Neurol. 2001, 16, 421–424. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.-E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Armeanu, R.; Mokkonen, M.; Crespi, B. Meta-analysis of BDNF levels in autism. Cell. Mol. Neurobiol. 2017, 37, 949–954. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Love, T.; Zhang, H. Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol. Psychiatry 2007, 61, 449–457. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 2017, 77, 393–404. [Google Scholar] [CrossRef]
- Bjorklund, G.; Saad, K.; Chirumbolo, S.; Kern, J.; Geier, D.; Geier, M.; Urbina, M. Immune dysfunction and neuroinflammation in autism spectrum disorder. Acta Neurobiol. Exp. 2016, 76, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.; Frye, R.; James, S. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Warren, R.; Averett, R.; Ghaziuddin, M. Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr. Neurol. 1997, 17, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.; Strunecka, A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 2009, 16, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Cohly, H.H.P.; Panja, A. Immunological findings in autism. Int. Rev. Neurobiol. 2005, 71, 317–341. [Google Scholar]
- Abdolmaleky, H.M.; Martin, M.; Zhou, J.-R.; Thiagalingam, S. Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases. Genes 2023, 14, 896. [Google Scholar] [CrossRef]
- Eftekharian, M.M.; Ghafouri-Fard, S.; Noroozi, R.; Omrani, M.D.; Arsang-Jang, S.; Ganji, M.; Gharzi, V.; Noroozi, H.; Komaki, A.; Mazdeh, M. Cytokine profile in autistic patients. Cytokine 2018, 108, 120–126. [Google Scholar] [CrossRef]
- Muratore, C.R.; Hodgson, N.W.; Trivedi, M.S.; Abdolmaleky, H.M.; Persico, A.M.; Lintas, C.; De La Monte, S.; Deth, R.C. Age-dependent decrease and alternative splicing of methionine synthase mRNA in human cerebral cortex and an accelerated decrease in autism. PLoS ONE 2013, 8, e56927. [Google Scholar] [CrossRef]
- Deth, R.C. Autism: A redox/methylation disorder. Glob. Adv. Health Med. 2013, 2, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Abdolmaleky, H.M.; Zhou, J.R. Microbiome, inflammation, epigenetic alterations, and mental diseases. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Nohesara, S.; Abdolmaleky, H.M.; Thiagalingam, S. Epigenetic Aberrations in Major Psychiatric Diseases Related to Diet and Gut Microbiome Alterations. Genes 2023, 14, 1506. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.J.; Tsuang, M.T.; Winn, M.; Chandler, S.D.; Collins, M.; Lopez, L.; Weinfeld, M.; Carter, C.; Schork, N.; Pierce, K. Blood-based gene expression signatures of infants and toddlers with autism. J. Am. Acad. Child Adolesc. Psychiatry 2012, 51, 934–944.e2. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.; Al-Ayadhi, L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J. Neuroinflamm. 2014, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis. 2008, 30, 303–311. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Snow, A.V.; Stary, J.M.; Araghi-Niknam, M.; Reutiman, T.J.; Lee, S.; Brooks, A.I.; Pearce, D.A. Reelin signaling is impaired in autism. Biol. Psychiatry 2005, 57, 777–787. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Zhubi, A.; Chen, Y.; Guidotti, A.; Grayson, D.R. Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. Int. J. Dev. Neurosci. 2017, 62, 63–72. [Google Scholar] [CrossRef]
- Samanta, D. An updated review of tuberous sclerosis complex-associated autism spectrum disorder. Pediatr. Neurol. 2020, 109, 4–11. [Google Scholar] [CrossRef]
- Mejia, L.A.; Litterman, N.; Ikeuchi, Y.; de la Torre-Ubieta, L.; Bennett, E.J.; Zhang, C.; Harper, J.W.; Bonni, A. A novel Hap1–Tsc1 interaction regulates neuronal mTORC1 signaling and morphogenesis in the brain. J. Neurosci. 2013, 33, 18015–18021. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Gower, A.C.; Wong, C.K.; Cox, J.W.; Zhang, X.; Thiagalingam, A.; Shafa, R.; Sivaraman, V.; Zhou, J.R.; Thiagalingam, S. Aberrant transcriptomes and DNA methylomes define pathways that drive pathogenesis and loss of brain laterality/asymmetry in schizophrenia and bipolar disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2019, 180, 138–149. [Google Scholar] [CrossRef]
- Islam, M.M.; Zhang, C.-L. TLX: A master regulator for neural stem cell maintenance and neurogenesis. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 210–216. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, H.; Ma, G.; Cao, X.; Liu, Z. Reelin is involved in transforming growth factor-β1-induced cell migration in esophageal carcinoma cells. PLoS ONE 2012, 7, e31802. [Google Scholar] [CrossRef] [PubMed]
- Genestine, M.; Lin, L.; Durens, M.; Yan, Y.; Jiang, Y.; Prem, S.; Bailoor, K.; Kelly, B.; Sonsalla, P.K.; Matteson, P.G. Engrailed-2 (En2) deletion produces multiple neurodevelopmental defects in monoamine systems, forebrain structures and neurogenesis and behavior. Hum. Mol. Genet. 2015, 24, 5805–5827. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Bajetto, A.; Barbero, S.; Pirani, P.; Florio, T.; Schettini, G. Chemokines and their receptors in the CNS: Expression of CXCL12/SDF-1 and CXCR4 and their role in astrocyte proliferation. Toxicol. Lett. 2003, 139, 181–189. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. An update on the epigenetics of psychotic diseases and autism. Epigenomics 2015, 7, 427–449. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. Cataloging recent advances in epigenetic alterations in major mental disorders and autism. Epigenomics 2021, 13, 1231–1245. [Google Scholar] [CrossRef]
- Stover, A.E.; Brick, D.J.; Nethercott, H.E.; Banuelos, M.G.; Sun, L.; O’Dowd, D.K.; Schwartz, P.H. Process-based expansion and neural differentiation of human pluripotent stem cells for transplantation and disease modeling. J. Neurosci. Res. 2013, 91, 1247–1262. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 31241. [Google Scholar] [CrossRef]
- Redcay, E.; Courchesne, E. When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol. Psychiatry 2005, 58, 1–9. [Google Scholar] [CrossRef]
- Correia, C.; Coutinho, A.; Sequeira, A.; Sousa, I.; Lourenco Venda, L.; Almeida, J.; Abreu, R.; Lobo, C.; Miguel, T.; Conroy, J. Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav. 2010, 9, 841–848. [Google Scholar] [CrossRef]
- Chandley, M.J.; Crawford, J.D.; Szebeni, A.; Szebeni, K.; Ordway, G.A. NTRK2 expression levels are reduced in laser captured pyramidal neurons from the anterior cingulate cortex in males with autism spectrum disorder. Mol. Autism 2015, 6, 28. [Google Scholar] [CrossRef]
- Bundo, M.; Ueda, J.; Nakachi, Y.; Kasai, K.; Kato, T.; Iwamoto, K. Decreased DNA methylation at promoters and gene-specific neuronal hypermethylation in the prefrontal cortex of patients with bipolar disorder. Mol. Psychiatry 2021, 26, 3407–3418. [Google Scholar] [CrossRef]
- Storer, M.A.; Gallagher, D.; Fatt, M.P.; Simonetta, J.V.; Kaplan, D.R.; Miller, F.D. Interleukin-6 regulates adult neural stem cell numbers during normal and abnormal post-natal development. Stem Cell Rep. 2018, 10, 1464–1480. [Google Scholar] [CrossRef]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.-M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef]
- Basheer, S.; Venkataswamy, M.M.; Christopher, R.; Van Amelsvoort, T.; Srinath, S.; Girimaji, S.C.; Ravi, V. Immune aberrations in children with autism spectrum disorder: A case-control study from a tertiary care neuropsychiatric hospital in India. Psychoneuroendocrinology 2018, 94, 162–167. [Google Scholar] [CrossRef]
- Cao, X.; Liu, K.; Liu, J.; Liu, Y.-W.; Xu, L.; Wang, H.; Zhu, Y.; Wang, P.; Li, Z.; Wen, J. Dysbiotic gut microbiota and dysregulation of cytokine profile in children and teens with autism spectrum disorder. Front. Neurosci. 2021, 15, 635925. [Google Scholar] [CrossRef]
- Aigner, L.; Wachs, F.; Winner, B.; Couillard-Despres, S.; Schiller, T.; Aigner, R.; Winkler, J.; Bogdahn, U. Transforming growth factor-beta1 is a negative modulator of adult neurogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 358–370. [Google Scholar]
- Romão, L.F.; de Oliveira Sousa, V.; Neto, V.M.; Gomes, F.C.A. Glutamate activates GFAP gene promoter from cultured astrocytes through TGF-β1 pathways. J. Neurochem. 2008, 106, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, Y.; Sousa, N.; Almeida, O.F. SMAD pathway mediation of BDNF and TGFβ2 regulation of proliferation and differentiation of hippocampal granule neurons. Development 2005, 132, 3231–3242. [Google Scholar] [CrossRef] [PubMed]
- Folsom, T.D.; Fatemi, S.H. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013, 68, 122–135. [Google Scholar] [CrossRef]
- Lee, H.-J.; Park, J.-H.; Trotter, J.H.; Maher, J.N.; Keenoy, K.E.; Jang, Y.M.; Lee, Y.; Kim, J.-I.; Weeber, E.J.; Hoe, H.-S. Reelin and APP Cooperatively Modulate Dendritic Spine Formation In Vitro and In Vivo. Exp. Neurobiol. 2023, 32, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, C.; Yu, H.; Zhang, W.; Cheng, F.; Yu, H.; Zhou, D.; Li, B.; Liu, J.; Dai, J. Association between the methylation of six apoptosis-associated genes with autism spectrum disorder. Mol. Med. Rep. 2018, 18, 4629–4634. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.-H.; Zhang, W.; Jian, X. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Nestor, C.E.; Ottaviano, R.; Reddington, J.; Sproul, D.; Reinhardt, D.; Dunican, D.; Katz, E.; Dixon, J.M.; Harrison, D.J.; Meehan, R.R. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012, 22, 467–477. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011, 25, 2436–2452. [Google Scholar] [CrossRef]
- Kassai, H.; Sugaya, Y.; Noda, S.; Nakao, K.; Maeda, T.; Kano, M.; Aiba, A. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014, 7, 1626–1639. [Google Scholar] [CrossRef]
- Curatolo, P.; Scheper, M.; Emberti Gialloreti, L.; Specchio, N.; Aronica, E. Is tuberous sclerosis complex-associated autism a preventable and treatable disorder? World J. Pediatr. 2023, 20, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Koeglsperger, T.; Li, S.; Brenneis, C.; Saulnier, J.L.; Mayo, L.; Carrier, Y.; Selkoe, D.J.; Weiner, H.L. Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-β1 in the CNS. Glia 2013, 61, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Role of glutamate transporters in the pathophysiology of major mental illnesses. Nihon Shinkei Seishin Yakurigaku Zasshi Jpn. J. Psychopharmacol. 2009, 29, 161–164. [Google Scholar]
- Xu, S.; Han, J.; Morales, A.; Menzie, C.; Williams, K.; Fan, Y.-S. Characterization of 11p14-p12 deletion in WAGR syndrome by array CGH for identifying genes contributing to mental retardation and autism. Cytogenet. Genome Res. 2008, 122, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Aida, T.; Yoshida, J.; Nomura, M.; Tanimura, A.; Iino, Y.; Soma, M.; Bai, N.; Ito, Y.; Cui, W.; Aizawa, H. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology 2015, 40, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Allritz, C.; Engele, J.; Rein, T. DNA methylation dependent silencing of the human glutamate transporter EAAT2 gene in glial cells. Glia 2007, 55, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.-F.; Choi, Y.; Ayers-Ringler, J.R.; Biernacka, J.M.; Geske, J.R.; Lindberg, D.R.; McElroy, S.L.; Frye, M.A.; Choi, D.-S.; Veldic, M. Differential SLC1A2 promoter methylation in bipolar disorder with or without addiction. Front. Cell. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Dong, Q.; Yao, H.; Zhang, Y.; Yang, Y.; Dang, Y.; Zhang, H.; Yang, Z.; Xu, M.; Xu, R. Induced neural stem cells modulate microglia activation states via CXCL12/CXCR4 signaling. Brain Behav. Immun. 2017, 59, 288–299. [Google Scholar] [CrossRef]
- Wang, R.; Wu, Y.; Liu, R.; Liu, M.; Li, Q.; Ba, Y.; Huang, H. Deciphering therapeutic options for neurodegenerative diseases: Insights from SIRT1. J. Mol. Med. 2022, 100, 537–553. [Google Scholar] [CrossRef]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Kim, S.M.; Cho, S.Y.; Kim, M.W.; Roh, S.R.; Shin, H.S.; Suh, Y.H.; Geum, D.; Lee, M.A. Genome-wide analysis identifies NURR1-controlled network of new synapse formation and cell cycle arrest in human neural stem cells. Mol. Cells 2020, 43, 551. [Google Scholar]
- Popichak, K.A.; Hammond, S.L.; Moreno, J.A.; Afzali, M.F.; Backos, D.S.; Slayden, R.D.; Safe, S.; Tjalkens, R.B. Compensatory expression of Nur77 and Nurr1 regulates NF-κB–dependent inflammatory signaling in astrocytes. Mol. Pharmacol. 2018, 94, 1174–1186. [Google Scholar] [CrossRef]
- Alam, M.M.; Yang, D.; Li, X.-Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef]
- Guarente, L. Diverse and dynamic functions of the Sir silencing complex. Nat. Genet. 1999, 23, 281–285. [Google Scholar] [CrossRef]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 2011, 9, 11. [Google Scholar] [CrossRef]
- Nardone, S.; Sharan Sams, D.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef]
- Irwin, J.L.; Yeates, A.J.; Mulhern, M.S.; McSorley, E.M.; Strain, J.; Watson, G.E.; Grzesik, K.; Thurston, S.W.; Love, T.M.; Smith, T.H. Maternal gestational immune response and autism spectrum disorder phenotypes at 7 years of age in the seychelles child development study. Mol. Neurobiol. 2019, 56, 5000–5008. [Google Scholar] [CrossRef]
- Leviton, A.; Joseph, R.M.; Allred, E.N.; Fichorova, R.N.; O’Shea, T.M.; Kuban, K.K.; Dammann, O. The risk of neurodevelopmental disorders at age 10 years associated with blood concentrations of interleukins 4 and 10 during the first postnatal month of children born extremely preterm. Cytokine 2018, 110, 181–188. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Fogo, M.; Pinto, B.; Alves, A.; Suzuki, A.; Morales, A.; Ezquina, S.; Sosa, O.; Sutton, G.; Sunaga-Franze, D. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol. Psychiatry 2021, 26, 1589–1605. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Autism | ||||||
|---|---|---|---|---|---|---|---|
| ID | Age/Year | Sex | Sources | ID | Age/Year | Sex | Sources |
| SV0000525 | 6 | Female | Simon’s foundation | SV0000540 | 14 | Male | Simon’s foundation |
| SS0013111 | 45 | Male | Simon’s foundation | SS0013099 | 14 | Male | Simon’s foundation |
| SV0001455 | 12 | Male | Simon’s foundation | SV0001473 | 6 | Female | Simon’s foundation |
| SV0001464 | 3 | Female | Simon’s foundation | SC171 | Unknown | Male × | NIMH |
| SC173 | Unknown | Male × | NIMH | SC126 | Fragile × | Male × | NIMH |
| Astrocyte | Neuron | Expression Ratio | |
|---|---|---|---|
| Gene | Fold change in autism (Mean ± SEM of controls/cases, 95% CI) t, p value | Fold change in autism (Mean ± SEM of controls/cases, 95% CI) t, p value | Astrocyte/neuron ratio in controls |
| TGFB1 | 2.3 (0.94 ± 0.17/2.3 ± 0.34, −2.2 to −0.54) t = 3.9, p = 0.006 | 1.5, not significant (NS) | 6 |
| TGFB2 | 2 (1 ± 0.17/2 ± 0.4, −2.197 to −0.3160) t = 3.2, p = 0.02 | 1.4 (NS) | >3 |
| TGFB3 | NS | NS | 2.2 |
| NR2E1 | 0.36 (1 ± 0.3/0.37 ± 0.07, −0.13 to 1.4) p = 0.09 | NS | >15 |
| IL4 | 0.21 (1.06 ± 0.31/0.17 ± 0.06, 0.05 to 1.7) t = 2.5, p = 0.04 | NS | 3.5 |
| IL6 | 34 (1.1 ± 0.3/37 ± 7.4, −51 to −20) t = 5.5, p = 0.001 | 28 (1 ± 0.16/28 ± 0.2.8) p = 0.053 | >30 |
| IL1B | NS | NS | 2 |
| TNFA | NS | No expression | N/A |
| TNFRSF1A | NS | NS | >1.5 |
| IFI16 | 1.5 (0.99 ± 0.12/1.5 ± 0.14, −0.97 to −0.12) t = 3, p = 0.02 | NS | ~7 |
| IGFBPL1 | 0.04 (1.1 ± 0.38/0.03 ± 0.01, −0.08 to 2) t = 2.5, p = 0.04 | NS | >2 |
| GPX1 | 0.6 (1 ± 0.1/0.6 ± 0.1) t = 3.2, p = 0.014 | 0.7 (1.1 ± 0.11/0.78 ± 0.13, −0.08 to 0.7) t = 1.9, p = 0.1 | 1 |
| SIRT1 | 0.32 (1.05 ± 0.1/0.32 ± 0.06, 0.44 to 1) t = 5.8, p = 0.0006 | NS | 1 |
| NURR1 | 0.1 (1 ± 0.3/0.07 ± 0.02, 0.25 to 1.6) t = 3.2, p = 0.01 | NS | 2 |
| SNCA | 0.4 (1 ± 0.1/0.4 ± 0.1, 0.27 to 0.94) t = 4.4, p = 0.004 | NS | 1.5 |
| BDNF | 1.8 (0.94 ± 0.2/1.8 ± 0.15, −1.5 to −0.26) t = 3.4, p = 0.01 | NS | 3 |
| CXCR4 | 0.2 (1 ± 0.3/0.18 ± 0.1, 0.3 to 1.3) t = 3.9, p = 0.006 | NS | 3 |
| EN2 | 0.2 (1 ± 0.25/0.24 ± 0.19, −0.01 to 1.5) t = 2.23, p = 0.052 | 3.3 (1 ± 0.3/3.6 ± 1.2, −5.2 to −0.002) t = 2.4, p = 0.05 | >20 |
| HAP1 | 0.25 (1 ± 0.08/0.25 ± 0.1, 0.52 to 1.1) t = 6.3, p = 0.0004 | 0.4 (1 ± 0.26/0.4 ± 0.14, −0.18 to 1.3) t = 1.8, p = 0.11 | 0.5 |
| RELN | 0.032 (1 ± 0.2/0.035 ± 0.02, 0.1 ± 0.16) t= 5.9, p = 0.0006 | NS | 0.5 |
| NTRK2 | 0.04 (1 ± 0.2/0.04 ± 0.01, 0.35 to 1.5) t = 3.8, p = 0.01 | NS | 2.9 |
| SLC1A2 | 0.11 (1.03 ± 0.35/0.1 ± 0.08, 0.07 to 1.75) t = 2.6, p = 0.045 | 0.5 (1 ± 0.24/0.5 ± 0.14, −0.27 to 1.141) t = 1.5, p = 0.18 | 2 |
| SLC1A3 | 0.11 (1 ± 0.15/0.11 ± 0.04, 0.45 to 1.3) t = 4.9, p = 0.002 | NS | >4 |
| SNAI1/Snail | NS | NS | 1.5 |
| SYN1 (synapsin) | N/A | 0.52 (1 ± 0.17/0.5 ± 0.17, −0.1 to 1) t = 1.9, p = 0.1 | 0.045 |
| Astrocytes | Neurons | |||
|---|---|---|---|---|
| Gene | 5-mc (Mean ± SEM Cont./Autism, 95% CI) t, p value | 5-hmc (Mean ± SEM Cont./Autism, 95% CI) t, p value | 5-mc (Mean ± SEM Cont./Autism, 95% CI) t, p value | 5-hmc (Mean ± SEM Cont./Autism, 95% CI) t, p value |
| TGFB1 | Unmethylated | Unmethylated | NS (no significant change) | NS |
| TGFB2 Site A | Hypomethylation (1 ± 0.1/0.58 ± 0.13, 0.1 to 0.7) t = 3.2, p = 0.04 | Negligible 5-hmc | NS | NS |
| TGFB2 Site B | Unmethylated | Unmethylated | Heavily methylated, 50% more in autism but NS | ~50% more in autism (1 ± 0.1/1.460 ± 0.11, 0.8 to −0.08), t = 2.9, p = 0.03 |
| IL6 | Hypomethylated (0.95 ± 0.17/0.46 ± 0.03, 0.02 to 1), t = 2.5, p = 0.04 | NS | NS | NS |
| TNFA | Hypomethylated (1 ± 0.2/0.34 ± 0.03, 0.07 to 1.2), t = 2.6, p = 0.03 | NS | NS | NS |
| IFI16 | No methylation | No 5-hmc | Hypomethylated (1 ± 0.14/0.48 ± 0.11, 0.08 to 0.96) t = 2.8, p = 0.02 | NS |
| EN2 | Hypomethylated (1 ± 0.15/0.53 ± 0.025, 0.09 to 0.86) t = 2.9, p = 0.02 | NS | NS | NS |
| HAP1 | NS | Decreased (1 ± 0.15/0.5 ± 0.05, 0.1 to 0.96) t = 2.9, p = 0.02 | Hypermethylated (1 ± 0.3/2.5 ± 0.5, −2.7 to −0.2) t= 2.7, p = 0.04 | NS |
| RELN | 10% methylation, NS | Negligible 5-hmc | NS | Negligible 5-hmc |
| SLC1A2 | Negligible methylation | Negligible 5-hmc | NS | Negligible 5-hmc |
| SLC1A3 | No methylation | No 5-hmc | Hypomethylated (1 ± 0.1/0.6 ± 0.11, 0.04 to 0.75) t = 2.6, p = 0.035 | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafavi Abdolmaleky, H.; Alam, R.; Nohesara, S.; Deth, R.C.; Zhou, J.-R. iPSC-Derived Astrocytes and Neurons Replicate Brain Gene Expression, Epigenetic, Cell Morphology and Connectivity Alterations Found in Autism. Cells 2024, 13, 1095. https://doi.org/10.3390/cells13131095
Mostafavi Abdolmaleky H, Alam R, Nohesara S, Deth RC, Zhou J-R. iPSC-Derived Astrocytes and Neurons Replicate Brain Gene Expression, Epigenetic, Cell Morphology and Connectivity Alterations Found in Autism. Cells. 2024; 13(13):1095. https://doi.org/10.3390/cells13131095
Chicago/Turabian StyleMostafavi Abdolmaleky, Hamid, Reza Alam, Shabnam Nohesara, Richard C. Deth, and Jin-Rong Zhou. 2024. "iPSC-Derived Astrocytes and Neurons Replicate Brain Gene Expression, Epigenetic, Cell Morphology and Connectivity Alterations Found in Autism" Cells 13, no. 13: 1095. https://doi.org/10.3390/cells13131095
APA StyleMostafavi Abdolmaleky, H., Alam, R., Nohesara, S., Deth, R. C., & Zhou, J.-R. (2024). iPSC-Derived Astrocytes and Neurons Replicate Brain Gene Expression, Epigenetic, Cell Morphology and Connectivity Alterations Found in Autism. Cells, 13(13), 1095. https://doi.org/10.3390/cells13131095