
Challenges of Regulated Cell Death: Implications for Therapy Resistance in Cancer


Abstract
1. Introduction
2. Regulated Cell Death Mechanisms
3. Regulated Non-Necrotic Cell Death and Implications in Drug Resistance
3.1. Apoptosis
3.1.1. Intrinsic Apoptosis
3.1.2. Extrinsic Apoptosis
3.1.3. Apoptosis and Drug Resistance
3.2. Autophagy-Dependent Cell Death
4. Regulated Necrotic Cell Death and Implications in Drug Resistance
4.1. Pyroptosis
4.2. Ferroptosis
4.3. Necroptosis
5. Other Modalities of Regulated Cell Death and Implications in Drug Resistance
5.1. Parthanatos
5.2. Cuproptosis
5.3. Disulfidptosis
5.4. Lysosome-Dependent Cell Death
5.5. Alkaliptosis
5.6. Entotic Cell Death
5.7. Immunogenic Cell Death
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liao, M.; Qin, R.; Huang, W.; Zhu, H.P.; Peng, F.; Han, B.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: A revisited perspective from molecular mechanisms to targeted therapies. J. Hematol. Oncol. 2022, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; De Luca, A.; Avena, P.; De Amicis, F.; Casaburi, I.; Sirianni, R.; Pezzi, V. Estrogen Receptors-Mediated Apoptosis in Hormone-Dependent Cancers. Int. J. Mol. Sci. 2022, 23, 1242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, R.; Law, J.H.; Khan, M.; Yip, K.W.; Sun, Q. Editorial: Hallmark of cancer: Resisting cell death. Front. Oncol. 2022, 12, 1069947. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; De Amicis, F. Aberrant Notch signaling in gliomas: A potential landscape of actionable converging targets for combination approach in therapies resistance. Cancer Drug Resist. 2022, 5, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Publisher Correction: Harnessing innate immunity in cancer therapy. Nature 2019, 576, E3. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wei, Z.; Fu, K.; Duan, Y.; Zhang, M.; Li, K.; Guo, T.; Yin, R. Non-apoptotic cell death in ovarian cancer: Treatment, resistance and prognosis. Biomed. Pharmacother. 2022, 150, 112929. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sun, C.; Li, Y.; Gao, X.; He, S.; Li, T.; Tian, D. Autophagy-related genes Raptor, Rictor, and Beclin1 expression and relationship with multidrug resistance in colorectal carcinoma. Hum. Pathol. 2015, 46, 1752–1759. [Google Scholar] [CrossRef]
- Chaudhary, N.; Choudhary, B.S.; Shah, S.G.; Khapare, N.; Dwivedi, N.; Gaikwad, A.; Joshi, N.; Raichanna, J.; Basu, S.; Gurjar, M.; et al. Lipocalin 2 expression promotes tumor progression and therapy resistance by inhibiting ferroptosis in colorectal cancer. Int. J. Cancer 2021, 149, 1495–1511. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Trovato-Salinaro, A.; Spampinato, G.; Musso, N.; Castorina, S.; Rizzarelli, E.; Condorelli, D.F. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation of SLC31A1,SCO1, and COX11 in colorectal cancer. FEBS Open Bio 2016, 6, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Weng, J.; Xiao, C.; Lu, J.; Cao, W.; Song, F.; He, Z.; Zhang, P.; Zhu, Z.; Xu, J. Cuproptosis-related molecular patterns and gene (ATP7A) in hepatocellular carcinoma and their relationships with tumor immune microenvironment and clinical features. Cancer Rep. 2023, 6, e1904. [Google Scholar] [CrossRef]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef] [PubMed]
- Takács-Vellai, K. Apoptosis and Autophagy, Different Modes of Cell Death: How to Utilize Them to Fight Diseases? Int. J. Mol. Sci. 2023, 24, 11609. [Google Scholar] [CrossRef]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. Regulated Necrotic Cell Death in Alternative Tumor Therapeutic Strategies. Cells 2020, 9, 2709. [Google Scholar] [CrossRef]
- Hadian, K.; Stockwell, B.R. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat. Rev. Drug Discov. 2023, 22, 723–742. [Google Scholar] [CrossRef]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondrial regulation of cell death: A phylogenetically conserved control. Microb. Cell 2016, 3, 101–108. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Kushnareva, Y.; Moraes, V.; Suess, J.; Peters, B.; Newmeyer, D.D.; Kuwana, T. Disruption of mitochondrial quality control genes promotes caspase-resistant cell survival following apoptotic stimuli. J. Biol. Chem. 2022, 298, 101835. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Khurana, A.; Navik, U.; Allawadhi, P.; Bharani, K.K.; Weiskirchen, R. Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics. Sci 2022, 4, 15. [Google Scholar] [CrossRef]
- Toscano, M.A.; Bianco, G.A.; Ilarregui, J.M.; Croci, D.O.; Correale, J.; Hernandez, J.D.; Zwirner, N.W.; Poirier, F.; Riley, E.M.; Baum, L.G.; et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 2007, 8, 825–834. [Google Scholar] [CrossRef]
- Furne, C.; Corset, V.; Hérincs, Z.; Cahuzac, N.; Hueber, A.O.; Mehlen, P. The dependence receptor DCC requires lipid raft localization for cell death signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4128–4133. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Tu, H.; Costa, M. XIAP’s Profile in Human Cancer. Biomolecules 2020, 10, 1493. [Google Scholar] [CrossRef]
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2011, 18, 538–548. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; D’Amico, M.; De Luca, A.; Conforti, F.L.; Pezzi, V.; De Amicis, F. Resveratrol, Epigallocatechin Gallate and Curcumin for Cancer Therapy: Challenges from Their Pro-Apoptotic Properties. Life 2023, 13, 261. [Google Scholar] [CrossRef]
- Augimeri, G.; Montalto, F.I.; Giordano, C.; Barone, I.; Lanzino, M.; Catalano, S.; Andò, S.; De Amicis, F.; Bonofiglio, D. Nutraceuticals in the Mediterranean Diet: Potential Avenues for Breast Cancer Treatment. Nutrients 2021, 13, 2557. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; D’Amico, M.; Pezzi, V.; De Amicis, F. Notch Signaling in Breast Tumor Microenvironment as Mediator of Drug Resistance. Int. J. Mol. Sci. 2022, 23, 6296. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Sussman, R.T.; Ricci, M.S.; Hart, L.S.; Sun, S.Y.; El-Deiry, W.S. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol. Ther. 2007, 6, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Galán, P.; Roué, G.; Villamor, N.; Campo, E.; Colomer, D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood 2007, 109, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Cetraro, P.; Plaza-Diaz, J.; MacKenzie, A.; Abadía-Molina, F. A Review of the Current Impact of Inhibitors of Apoptosis Proteins and Their Repression in Cancer. Cancers 2022, 14, 1671. [Google Scholar] [CrossRef]
- De Amicis, F.; Chimento, A.; Montalto, F.I.; Casaburi, I.; Sirianni, R.; Pezzi, V. Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1087. [Google Scholar] [CrossRef]
- Montinaro, A.; Walczak, H. Harnessing TRAIL-induced cell death for cancer therapy: A long walk with thrilling discoveries. Cell Death Differ. 2023, 30, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Wang, Y.; Zhou, X.; Seras, L.; Quax, W.; Demaria, M. Enhanced extrinsic apoptosis of therapy-induced senescent cancer cells using a death receptor 5 (DR5) selective agonist. Cancer Lett. 2022, 525, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bairey, O.; Zimra, Y.; Shaklai, M.; Okon, E.; Rabizadeh, E. Bcl-2, Bcl-X, Bax, and Bak expression in short- and long-lived patients with diffuse large B-cell lymphomas. Clin. Cancer Res. 1999, 5, 2860–2866. [Google Scholar] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.A.; Petrova, S.; Pound, J.D.; Voss, J.J.; Melville, L.; Paterson, M.; Farnworth, S.L.; Gallimore, A.M.; Cuff, S.; Wheadon, H.; et al. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr. Biol. 2015, 25, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Kisslinger, A.; Procaccini, C.; Paladino, S.; Oliviero, O.; de Amicis, F.; Faicchia, D.; Fasano, D.; Caputo, M.; Matarese, G.; et al. Convergent Effects of Resveratrol and PYK2 on Prostate Cells. Int. J. Mol. Sci. 2016, 17, 1542. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Lubas, M.; Lund, A.H. Emerging connections between RNA and autophagy. Autophagy 2017, 13, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.; Montalto, F.I.; Panno, M.L.; Andò, S.; De Amicis, F. A Notch inhibitor plus Resveratrol induced blockade of autophagy drives glioblastoma cell death by promoting a switch to apoptosis. Am. J. Cancer Res. 2021, 11, 5933–5950. [Google Scholar] [PubMed]
- Yu, X.; Zhou, Z.; Tang, S.; Zhang, K.; Peng, X.; Zhou, P.; Zhang, M.; Shen, L.; Yang, L. MDK induces temozolomide resistance in glioblastoma by promoting cancer stem-like properties. Am. J. Cancer Res. 2022, 12, 4825–4839. [Google Scholar] [PubMed]
- De Amicis, F.; Guido, C.; Santoro, M.; Giordano, F.; Donà, A.; Rizza, P.; Pellegrino, M.; Perrotta, I.; Bonofiglio, D.; Sisci, D.; et al. Ligand activated progesterone receptor B drives autophagy-senescence transition through a Beclin-1/Bcl-2 dependent mechanism in human breast cancer cells. Oncotarget 2016, 7, 57955–57969. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Leone, A.; Roca, M.S.; Lombardi, R.; Piezzo, M.; Caputo, R.; Ciardiello, C.; Costantini, S.; Bruzzese, F.; Sisalli, M.J.; et al. Inhibition of autophagy by chloroquine prevents resistance to PI3K/AKT inhibitors and potentiates their antitumor effect in combination with paclitaxel in triple negative breast cancer models. J. Transl. Med. 2022, 20, 290. [Google Scholar] [CrossRef]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef]
- Ferreira, P.M.P.; Sousa, R.W.R.; Ferreira, J.R.O.; Militão, G.C.G.; Bezerra, D.P. Chloroquine and hydroxychloroquine in antitumor therapies based on autophagy-related mechanisms. Pharmacol. Res. 2021, 168, 105582. [Google Scholar] [CrossRef]
- Lee, M.J.; Park, J.S.; Jo, S.B.; Joe, Y.A. Enhancing Anti-Cancer Therapy with Selective Autophagy Inhibitors by Targeting Protective Autophagy. Biomol. Ther. 2023, 31, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Chen, L.; Liu, J.; Zhong, Y.; Deng, L. The MicroRNA-Based Strategies to Combat Cancer Chemoresistance via Regulating Autophagy. Front. Oncol. 2022, 12, 841625. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yan, J.; Li, Z.; Yang, L.; Ju, F.; Sun, Y. Recent trends in emerging strategies for ferroptosis-based cancer therapy. Nanoscale Adv. 2023, 5, 1271–1290. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nature Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- Hsu, S.K.; Li, C.Y.; Lin, I.L.; Syue, W.J.; Chen, Y.F.; Cheng, K.C.; Teng, Y.N.; Lin, Y.H.; Yen, C.H.; Chiu, C.C. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics 2021, 11, 8813–8835. [Google Scholar] [CrossRef]
- Wang, Z.; Bao, A.; Liu, S.; Dai, F.; Gong, Y.; Cheng, Y. A Pyroptosis-Related Gene Signature Predicts Prognosis and Immune Microenvironment for Breast Cancer Based on Computational Biology Techniques. Front. Genet. 2022, 13, 801056. [Google Scholar] [CrossRef]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef]
- Wang, L.; Qin, X.; Liang, J.; Ge, P. Induction of Pyroptosis: A Promising Strategy for Cancer Treatment. Front. Oncol. 2021, 11, 635774. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef]
- Zhang, C.C.; Li, C.G.; Wang, Y.F.; Xu, L.H.; He, X.H.; Zeng, Q.Z.; Zeng, C.Y.; Mai, F.Y.; Hu, B.; Ouyang, D.Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Yang, Y.; Wang, G.; Wang, Z.; Liu, J.; Zhang, L.; Yu, Y. Pyroptosis-Related Gene Signature Predicts Prognosis and Indicates Immune Microenvironment Infiltration in Glioma. Front. Cell Dev. Biol. 2022, 10, 862493. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Wilkens, R.; Hoffrichter, A.; Kleinsimlinghaus, K.; Bohl, B.; Haag, C.; Lehmann, N.; Schmidt, M.; Muñoz Perez-Vico, E.; Wangemann, J.; Rehder, K.F.; et al. Diverse maturity-dependent and complementary anti-apoptotic brakes safeguard human iPSC-derived neurons from cell death. Cell Death Dis. 2022, 13, 887. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, D.H.; Jeong, S.Y.; Park, S.H.; Oh, S.C.; Park, Y.S.; Yu, J.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J. Cell Biochem. 2019, 120, 928–939. [Google Scholar] [CrossRef]
- Kim, J.-W.; Min, D.W.; Kim, D.; Kim, J.; Kim, M.J.; Lim, H.; Lee, J.-Y. GPX4 overexpressed non-small cell lung cancer cells are sensitive to RSL3-induced ferroptosis. Sci. Rep. 2023, 13, 8872. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Ma, T.; Du, J.; Zhang, Y.; Wang, Y.; Wang, B.; Zhang, T. GPX4-independent ferroptosis-a new strategy in disease’s therapy. Cell Death Discov. 2022, 8, 434. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Shibata, Y.; Yasui, H.; Higashikawa, K.; Miyamoto, N.; Kuge, Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE 2019, 14, e0225931. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef]
- Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; Ge, P. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem. Biophys. Res. Commun. 2019, 518, 590–597. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Wang, X.; Tian, H.; Wang, Y.; Jin, J.; Shan, Z.; Liu, Y.; Cai, Z.; Tong, X.; et al. Stem Cell Factor SOX2 Confers Ferroptosis Resistance in Lung Cancer via Upregulation of SLC7A11. Cancer Res. 2021, 81, 5217–5229. [Google Scholar] [CrossRef]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.Y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Q.; Zhuo, Q.; Zhu, Y.; Fan, G.; Liu, M.; Sun, Q.; Zhang, Z.; Liu, W.; Xu, W.; et al. Abrogation of ARF6 promotes RSL3-induced ferroptosis and mitigates gemcitabine resistance in pancreatic cancer cells. Am. J. Cancer Res. 2020, 10, 1182–1193. [Google Scholar]
- Jiang, Z.; Lim, S.O.; Yan, M.; Hsu, J.L.; Yao, J.; Wei, Y.; Chang, S.S.; Yamaguchi, H.; Lee, H.H.; Ke, B.; et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Investig. 2021, 131, e139434. [Google Scholar] [CrossRef]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar] [CrossRef]
- Chen, D.; Tong, J.; Yang, L.; Wei, L.; Stolz, D.B.; Yu, J.; Zhang, J.; Zhang, L. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc. Natl. Acad. Sci. USA 2018, 115, 3930–3935. [Google Scholar] [CrossRef]
- Moquin, D.M.; McQuade, T.; Chan, F.K. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Xie, J.; Tian, W.; Tang, Y.; Zou, Y.; Zheng, S.; Wu, L.; Zeng, Y.; Wu, S.; Xie, X.; Xie, X. Establishment of a Cell Necroptosis Index to Predict Prognosis and Drug Sensitivity for Patients with Triple-Negative Breast Cancer. Front. Mol. Biosci. 2022, 9, 834593. [Google Scholar] [CrossRef]
- Baik, J.Y.; Liu, Z.; Jiao, D.; Kwon, H.J.; Yan, J.; Kadigamuwa, C.; Choe, M.; Lake, R.; Kruhlak, M.; Tandon, M.; et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat. Commun. 2021, 12, 2666. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, M.; Mei, L.; Ruan, J.; Hu, Q.; Peng, J.; Su, H.; Liao, H.; Liu, S.; Liu, W.; et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget 2016, 7, 22219–22233. [Google Scholar] [CrossRef]
- Koo, G.B.; Morgan, M.J.; Lee, D.G.; Kim, W.J.; Yoon, J.H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef]
- Safferthal, C.; Rohde, K.; Fulda, S. Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene 2017, 36, 1487–1502. [Google Scholar] [CrossRef]
- Feldmann, F.; Schenk, B.; Martens, S.; Vandenabeele, P.; Fulda, S. Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget 2017, 8, 68208–68220. [Google Scholar] [CrossRef]
- Huang, C.Y.; Kuo, W.T.; Huang, Y.C.; Lee, T.C.; Yu, L.C. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622. [Google Scholar] [CrossRef]
- Dai, W.; Cheng, J.; Leng, X.; Hu, X.; Ao, Y. The potential role of necroptosis in clinical diseases (Review). Int. J. Mol. Med. 2021, 47, 89. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, L.; Tao, S.; Yao, Y.; Wang, Y.; Wei, Q.; Shao, A.; Deng, Y. Parthanatos and its associated components: Promising therapeutic targets for cancer. Pharmacol. Res. 2021, 163, 105299. [Google Scholar] [CrossRef]
- Huang, P.; Chen, G.; Jin, W.; Mao, K.; Wan, H.; He, Y. Molecular Mechanisms of Parthanatos and Its Role in Diverse Diseases. Int. J. Mol. Sci. 2022, 23, 7292. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef]
- Shen, S.M.; Guo, M.; Xiong, Z.; Yu, Y.; Zhao, X.Y.; Zhang, F.F.; Chen, G.Q. AIF inhibits tumor metastasis by protecting PTEN from oxidation. EMBO Rep. 2015, 16, 1563–1580. [Google Scholar] [CrossRef] [PubMed]
- Gallego, M.A.; Joseph, B.; Hemström, T.H.; Tamiji, S.; Mortier, L.; Kroemer, G.; Formstecher, P.; Zhivotovsky, B.; Marchetti, P. Apoptosis-inducing factor determines the chemoresistance of non-small-cell lung carcinomas. Oncogene 2004, 23, 6282–6291. [Google Scholar] [CrossRef]
- Feng, X.; Zhou, Y.; Proctor, A.M.; Hopkins, M.M.; Liu, M.; Koh, D.W. Silencing of Apoptosis-Inducing factor and poly(ADP-ribose) glycohydrolase reveals novel roles in breast cancer cell death after chemotherapy. Mol. Cancer 2012, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Boulos, J.C.; Klauck, S.M.; Efferth, T. The cardiac glycoside ZINC253504760 induces parthanatos-type cell death and G2/M arrest via downregulation of MEK1/2 phosphorylation in leukemia cells. Cell Biol. Toxicol. 2023, 39, 2971–2997. [Google Scholar] [CrossRef] [PubMed]
- Cobine, P.A.; Brady, D.C. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol. Cell 2022, 82, 1786–1787. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Wang, Y.; Jiang, P.; Liu, F.; Feng, N. Ferredoxin 1 is a cuproptosis-key gene responsible for tumor immunity and drug sensitivity: A pan-cancer analysis. Front. Pharmacol. 2022, 13, 938134. [Google Scholar] [CrossRef]
- Dreishpoon, M.B.; Bick, N.R.; Petrova, B.; Warui, D.M.; Cameron, A.; Booker, S.J.; Kanarek, N.; Golub, T.R.; Tsvetkov, P. FDX1 regulates cellular protein lipoylation through direct binding to LIAS. bioRxiv 2023. [Google Scholar] [CrossRef]
- Safi, R.; Nelson, E.R.; Chitneni, S.K.; Franz, K.J.; George, D.J.; Zalutsky, M.R.; McDonnell, D.P. Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 2014, 74, 5819–5831. [Google Scholar] [CrossRef]
- Blockhuys, S.; Wittung-Stafshede, P. Roles of Copper-Binding Proteins in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 871. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Wang, F.; Lin, H.; Su, Q.; Li, C. Cuproptosis-related lncRNA predict prognosis and immune response of lung adenocarcinoma. World J. Surg. Oncol. 2022, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Steinbrueck, A.; Sedgwick, A.C.; Brewster, J.T., 2nd; Yan, K.C.; Shang, Y.; Knoll, D.M.; Vargas-Zúñiga, G.I.; He, X.P.; Tian, H.; Sessler, J.L. Transition metal chelators, pro-chelators, and ionophores as small molecule cancer chemotherapeutic agents. Chem. Soc. Rev. 2020, 49, 3726–3747. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.; Khan, A.A.; Aman, S.; Qamar, W.; Ebaid, H.; Al-Tamimi, J.; Alhazza, I.M.; Rady, A.M. Restrained management of copper level enhances the antineoplastic activity of imatinib in vitro and in vivo. Sci. Rep. 2018, 8, 1682. [Google Scholar] [CrossRef] [PubMed]
- Falls-Hubert, K.C.; Butler, A.L.; Gui, K.; Anderson, M.; Li, M.; Stolwijk, J.M.; Rodman, S.N., 3rd; Solst, S.R.; Tomanek-Chalkley, A.; Searby, C.C.; et al. Disulfiram causes selective hypoxic cancer cell toxicity and radio-chemo-sensitization via redox cycling of copper. Free Radic. Biol. Med. 2020, 150, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, L.; Zhang, Y.; Yan, Y.; Wang, C.; Colic, M.; Olszewski, K.; Horbath, A.; Chen, X.; Lei, G.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, J.; Liu, X.; Xiao, Z.; Lei, G.; Lee, H.; Koppula, P.; Cheng, W.; Mao, C.; Zhuang, L.; et al. H2A Monoubiquitination Links Glucose Availability to Epigenetic Regulation of the Endoplasmic Reticulum Stress Response and Cancer Cell Death. Cancer Res. 2020, 80, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Teng, H.; Hang, Q.; Kondiparthi, L.; Lei, G.; Horbath, A.; Liu, X.; Mao, C.; Wu, S.; Zhuang, L.; et al. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nat. Commun. 2023, 14, 3673. [Google Scholar] [CrossRef]
- Liu, X.; Gan, B. Glucose starvation induces NADPH collapse and disulfide stress in SLC7A11(high) cancer cells. Oncotarget 2021, 12, 1629–1630. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Meng, Y.; Dian, Y.; Zhou, Q.; Sun, Y.; Le, J.; Zeng, F.; Chen, X.; He, Y.; Deng, G. Molecular landmarks of tumor disulfidptosis across cancer types to promote disulfidptosis-target therapy. Redox Biol. 2023, 68, 102966. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Olsen, O.D.; Jäättelä, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [PubMed]
- Domagala, A.; Fidyt, K.; Bobrowicz, M.; Stachura, J.; Szczygiel, K.; Firczuk, M. Typical and Atypical Inducers of Lysosomal Cell Death: A Promising Anticancer Strategy. Int. J. Mol. Sci. 2018, 19, 2256. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhu, S.; Xie, Y.; Liu, J.; Sun, L.; Zeng, D.; Wang, P.; Ma, X.; Kroemer, G.; Bartlett, D.L.; et al. JTC801 Induces pH-dependent Death Specifically in Cancer Cells and Slows Growth of Tumors in Mice. Gastroenterology 2018, 154, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Liu, J.; Kang, R.; Yang, M.; Tang, D. Targeting NF-κB-dependent alkaliptosis for the treatment of venetoclax-resistant acute myeloid leukemia cells. Biochem. Biophys. Res. Commun. 2021, 562, 55–61. [Google Scholar] [CrossRef]
- Mlynarczuk-Bialy, I.; Dziuba, I.; Sarnecka, A.; Platos, E.; Kowalczyk, M.; Pels, K.K.; Wilczynski, G.M.; Wojcik, C.; Bialy, L.P. Entosis: From Cell Biology to Clinical Cancer Pathology. Cancers 2020, 12, 2481. [Google Scholar] [CrossRef]
- Santagostino, S.F.; Assenmacher, C.A.; Tarrant, J.C.; Adedeji, A.O.; Radaelli, E. Mechanisms of Regulated Cell Death: Current Perspectives. Vet. Pathol. 2021, 58, 596–623. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cibas, E.S.; Huang, H.; Hodgson, L.; Overholtzer, M. Induction of entosis by epithelial cadherin expression. Cell Res. 2014, 24, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, I.; Cantelli, G.; Bruce, F.; Sanz-Moreno, V. Rho, ROCK and actomyosin contractility in metastasis as drug targets. F1000Res 2016, 5, 783. [Google Scholar] [CrossRef] [PubMed]
- Hinojosa, L.S.; Holst, M.; Baarlink, C.; Grosse, R. MRTF transcription and Ezrin-dependent plasma membrane blebbing are required for entotic invasion. J. Cell Biol. 2017, 216, 3087–3095. [Google Scholar] [CrossRef] [PubMed]
- Kianfar, M.; Balcerak, A.; Chmielarczyk, M.; Tarnowski, L.; Grzybowska, E.A. Cell Death by Entosis: Triggers, Molecular Mechanisms and Clinical Significance. Int. J. Mol. Sci. 2022, 23, 4985. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Florey, O. Cancer cell cannibalism: Multiple triggers emerge for entosis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Zhang, Y.; Li, S.; Sun, F.; Wang, G.; Yang, T.; Wei, D.; Guo, L.; Xiao, H. Induction of entosis in prostate cancer cells by nintedanib and its therapeutic implications. Oncol. Lett. 2019, 17, 3151–3162. [Google Scholar] [CrossRef]
- Song, J.; Xu, R.; Zhang, H.; Xue, X.; Ruze, R.; Chen, Y.; Yin, X.; Wang, C.; Zhao, Y. Cell-in-Cell-Mediated Entosis Reveals a Progressive Mechanism in Pancreatic Cancer. Gastroenterology 2023, 165, 1505–1521.e1520. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Kielbik, M.; Szulc-Kielbik, I.; Klink, M. Calreticulin-Multifunctional Chaperone in Immunogenic Cell Death: Potential Significance as a Prognostic Biomarker in Ovarian Cancer Patients. Cells 2021, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, J.A.; Penton, K.E.; Zhao, Z.; Hayes, M.J.; Lima, S.M.; Irish, J.M.; Bachmann, B.O. An immunogenic cell injury module for the single-cell multiplexed activity metabolomics platform to identify promising anti-cancer natural products. J. Biol. Chem. 2022, 298, 102300. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of HMGB1-deficient tumors: Compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69–78. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Balogh, K.N.; Templeton, D.J.; Cross, J.V. Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses. PLoS ONE 2018, 13, e0197702. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Vanpouille-Box, C. Type I interferon induces cancer stem cells-mediated chemotherapy resistance. Oncoimmunology 2022, 11, 2127274. [Google Scholar] [CrossRef]
- Amiri, M.; Molavi, O.; Sabetkam, S.; Jafari, S.; Montazersaheb, S. Stimulators of immunogenic cell death for cancer therapy: Focusing on natural compounds. Cancer Cell Int. 2023, 23, 200. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Tohme, R.; Goldfinger, M.; Tomlinson, B.K.; Sakre, N.; Hanmer, S.; Jha, B.K.; Maciejewski, J.P.; Verma, A.; Saunthararajah, Y. Venetoclax Inhibition of Pyrimidine Synthesis Guides Methods for Integration with Decitabine or 5-Azacytidine That Are Non-Myelosuppressive. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Franzese, O.; Graziani, G. Role of PARP Inhibitors in Cancer Immunotherapy: Potential Friends to Immune Activating Molecules and Foes to Immune Checkpoints. Cancers 2022, 14, 5633. [Google Scholar] [CrossRef] [PubMed]
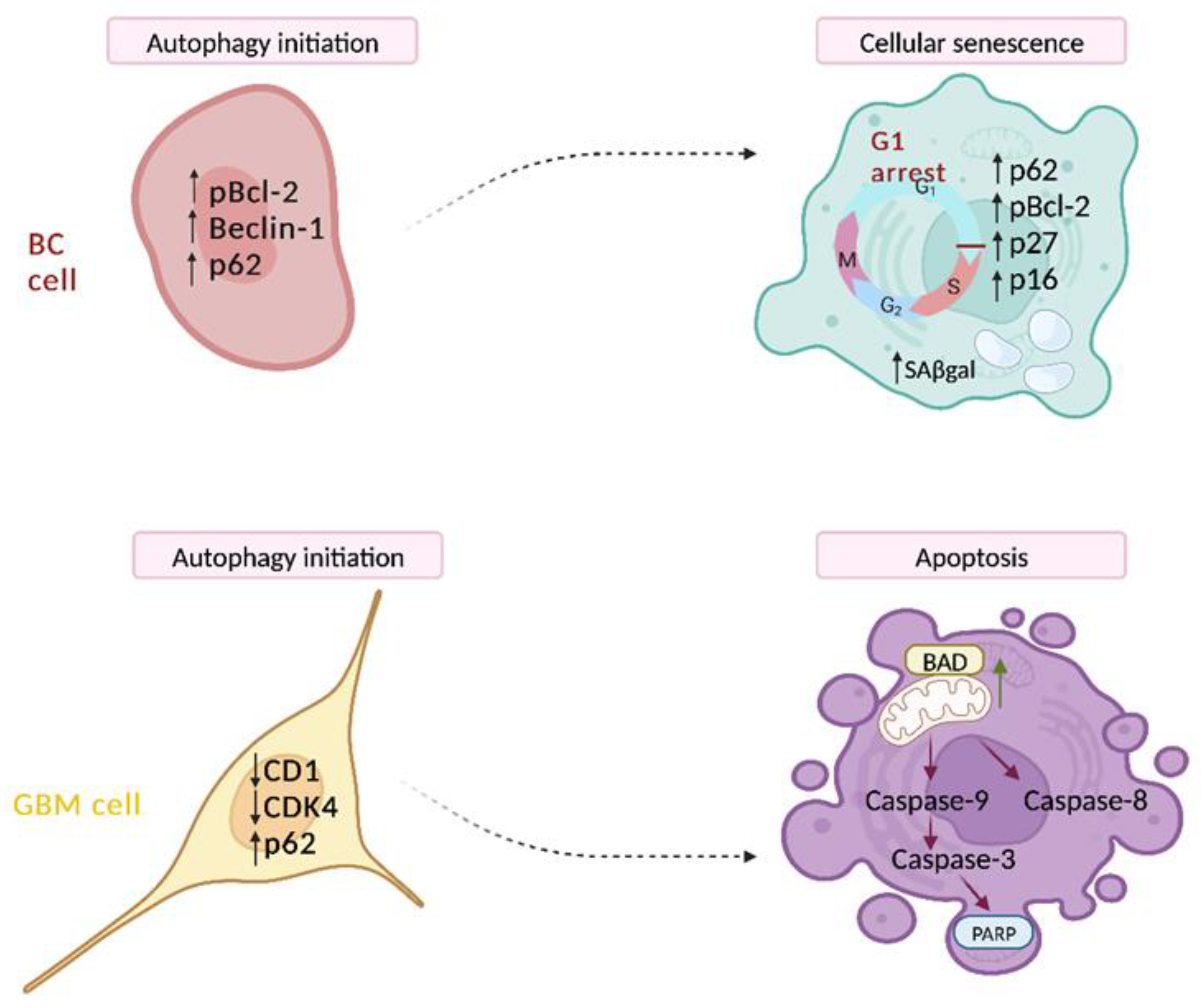


{kind=link}
{kind=link}
| Apoptosis | Pyroptosis | Ferroptosis | Autophagy | Entosis | Necroptosis | Parthanatos | ICD | |
|---|---|---|---|---|---|---|---|---|
| Pore forming protein | BAX/BAK (intrinsic apoptosis) | GSDMD GSDMB GSDME | Mitochondrial Permeability Transition Pore, ninjurin-1 | BID | Not present | Mixed lineage kinase domain-like protein (MLKL) | Ninjurin-1 | Not present |
| Morphologic hallmarks | Shrinkage of the cell, fragmentation into membrane-bound apoptotic bodies | Rapid loss of plasma membrane integrity; cell swelling and rupture | Cell swelling, plasma membrane rupture, smaller mitochondria | Autophagosomes, autolysosome | Cell-in-cell formation | Plasma membrane breakdown, releasing DAMPs | Loss of cell membrane integrity, mitochondrial abnormalities, nuclear shrinkage, and chromatin condensation | DAMPs; global arrest in Transcription and translation |
| Biochemical hallmarks or Biomarkers | Internucleosomal DNA fragmentation | Phosphatidylserine exposure and caspase activation | Iron accumulation, increased lipid peroxidation | ULK1 complex formation; turnover rate of the ATG8, LC3, p62 | Lipidation of LC3 onto the entotic vacuole | Phosphorylation of RIP1, RIP3, and MLKL | DNA fragmentations | Release of type I IFNs, ATP secretion, HMGB1, CALR, and other ER chaperone exposure |
| Inflammatory nature | no | yes | yes | yes | no | yes | no | yes |
| Molecule | Specific Action in Regulated Cell Death | Observations in Cancer Drug Resistance | Type of Cancer Models | Reference |
|---|---|---|---|---|
| Venetoclax (ABT-199) | Apoptosis inducer | ABT-199 inhibits the de novo pyrimidine synthesis enzyme, leading to overcoming the resistance against hypomethylating agents | Chronic lymphocytic leukemia, follicular lymphoma | [170] |
| Chloroquine (CQ) Hydroxychloroquine | Autophagy inhibitor | Used in combination with various anticancer drugs to enhance their cytotoxic effects and sensitize refractory cancers. | Central nervous system, lungs, breast, pancreas, leukocytes, skin, and colon/rectum cancers | [60] |
| Chloroquine (CQ) Hydroxychloroquine | Autophagy inhibitor | Chronic use of CQ has shown to overcome mechanism of drug resistance to PI3K/AKT inhibitors plus paclitaxel | Triple negative breast cancer | [56] |
| Tyrosine kinase inhibitor Nintedanib | Entosis induction | To inhibit cell proliferation and decrease the growth of xenografts | Prostate cancer | [157] |
| Multitargeting kinase inhibitor (sorafenib) | Necroptosis inhibitor | To restrict SMAC mimetic-induced necroptosis in apoptosis-resistant cells | Acute myeloid leukemia | [114] |
| PARP1 inhibitors (olaparib, rucaparib, niraparib and talazoparib) | Parthanatos inhibitors | Anti-tumor efficacy as monotherapy | Tumors expressing either germline or somatic mutations in the BRCA genes, advanced/metastatic ovarian cancer, triple negative breast cancer, pancreatic and prostate cancer | [171] |
| Molecule | Specific Action in Regulated Cell Death | Type of Cancer Models | Reference |
|---|---|---|---|
| 5-fluorouracil | Ferroptosis inhibitor | Colon cancer | [11] |
| BH3-mimetics: Venetoclax (ABT-199) | Apoptosis inducer | Chronic lymphocytic leukemia, follicular lymphoma | [170] |
| anti-PD-1 (mAbs) | Ferroptosis inducer | Melanoma, lung cancer, metastatic triple negative breast cancer, hepatocellular carcinoma | [102] |
| Niraparib | Parthanatos inhibitor | Tumors expressing either germline or somatic mutations in the BRCA genes, advanced/metastatic ovarian cancer, triple negative breast cancer, pancreatic and prostate cancer | [171] |
| Rucaparib | Parthanatos inhibitor | Tumors expressing either germline or somatic mutations in the BRCA genes, advanced/metastatic ovarian cancer, triple negative breast cancer, pancreatic and prostate cancer | [171] |
| Talazoparib | Parthanatos inhibitor | Tumors expressing either germline or somatic mutations in the BRCA genes, advanced/metastatic ovarian cancer, triple negative breast cancer, pancreatic and prostate cancer | [171] |
| Olaparib | Parthanatos inhibitor | Tumors expressing either germline or somatic mutations in the BRCA genes, advanced/metastatic ovarian cancer, triple negative breast cancer, pancreatic and prostate cancer | [171] |
| Nintedanib | Entosis inducer | Prostate cancer | [157] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, M.; De Amicis, F. Challenges of Regulated Cell Death: Implications for Therapy Resistance in Cancer. Cells 2024, 13, 1083. https://doi.org/10.3390/cells13131083
D’Amico M, De Amicis F. Challenges of Regulated Cell Death: Implications for Therapy Resistance in Cancer. Cells. 2024; 13(13):1083. https://doi.org/10.3390/cells13131083
Chicago/Turabian StyleD’Amico, Maria, and Francesca De Amicis. 2024. "Challenges of Regulated Cell Death: Implications for Therapy Resistance in Cancer" Cells 13, no. 13: 1083. https://doi.org/10.3390/cells13131083
APA StyleD’Amico, M., & De Amicis, F. (2024). Challenges of Regulated Cell Death: Implications for Therapy Resistance in Cancer. Cells, 13(13), 1083. https://doi.org/10.3390/cells13131083