

Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Single-Cell Data Retrieval and Bioinformatics
2.2. Discriminant Analyses
2.3. CRISPR-Cas9 Screen Data Retrieval
2.4. Protein–Protein Interaction (PPI)
2.5. Survival Analysis
2.6. RNA-Seq Data Analysis and Bioinformatics
2.7. Cell Culture and Gene Knockdown
2.8. Colony Forming Unit (CFU) Assay and Detection of Cell Death Using Fluorescence Microscopy
2.9. Scratch Assay
2.10. Organoid Dome Culture
2.11. Statistical Analysis
3. Results
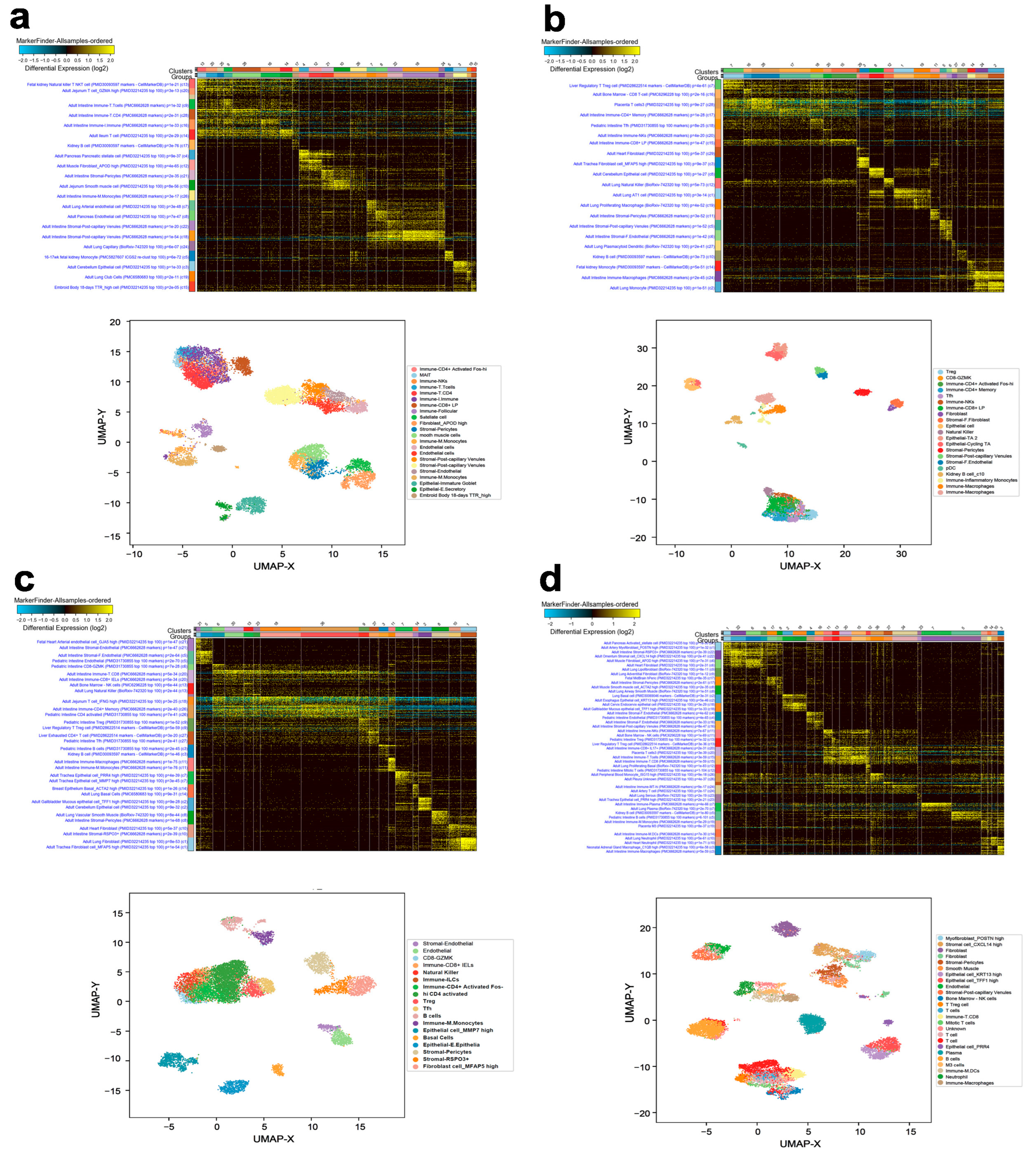
3.1. Single-Cell Transcriptome Analysis Revealed the Cellular Compositions of Different BC Molecular Subtypes
3.2. Identification of Unique Gene Markers Expressed by Cancerous Epithelial Cells in Each BC Molecular Subtype
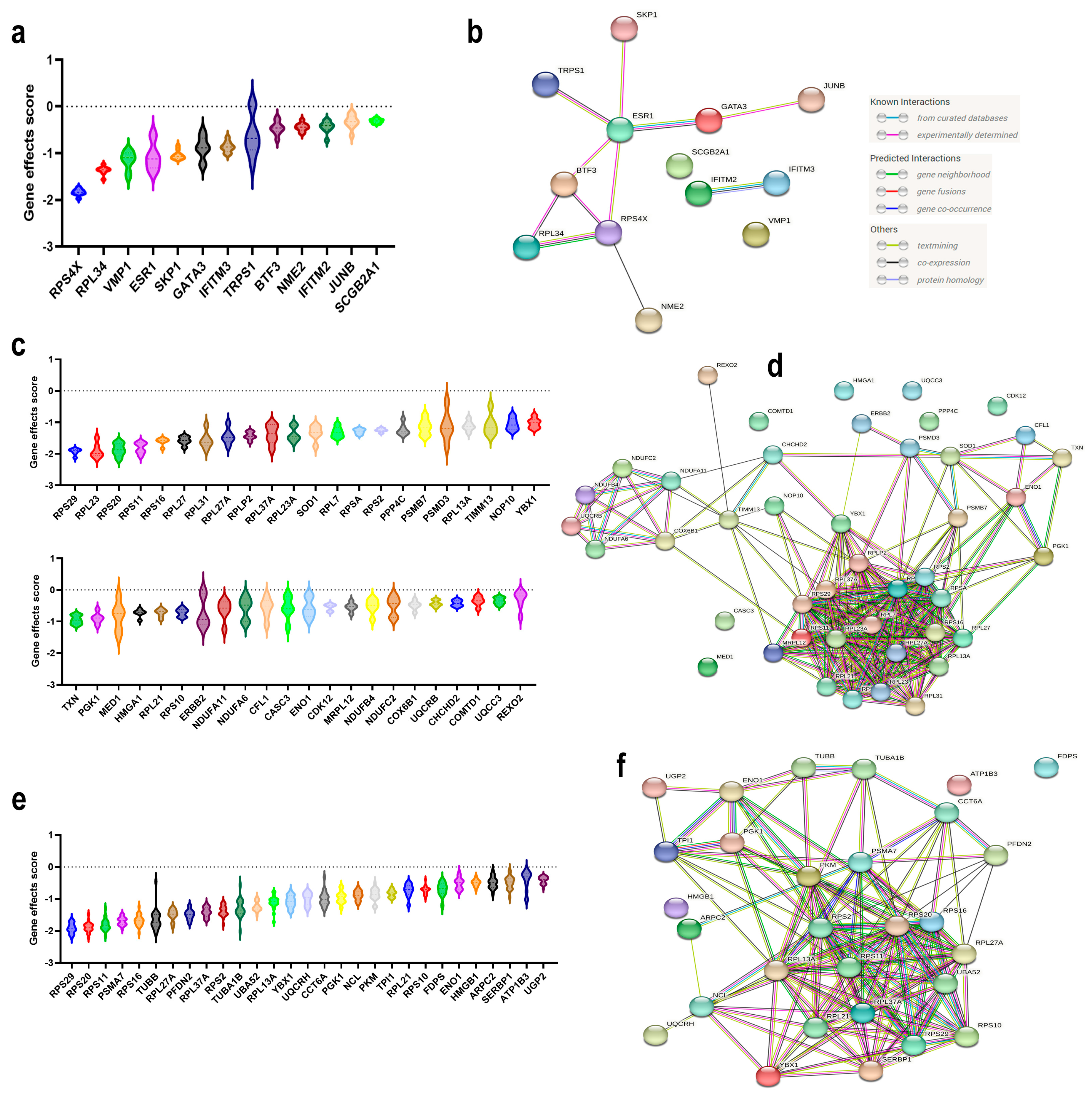
3.3. Identification of Novel Therapeutic Gene Targets for Each BC Molecular Subtype
3.4. Prognostic Value of the Identified TNBC Therapeutic Targets
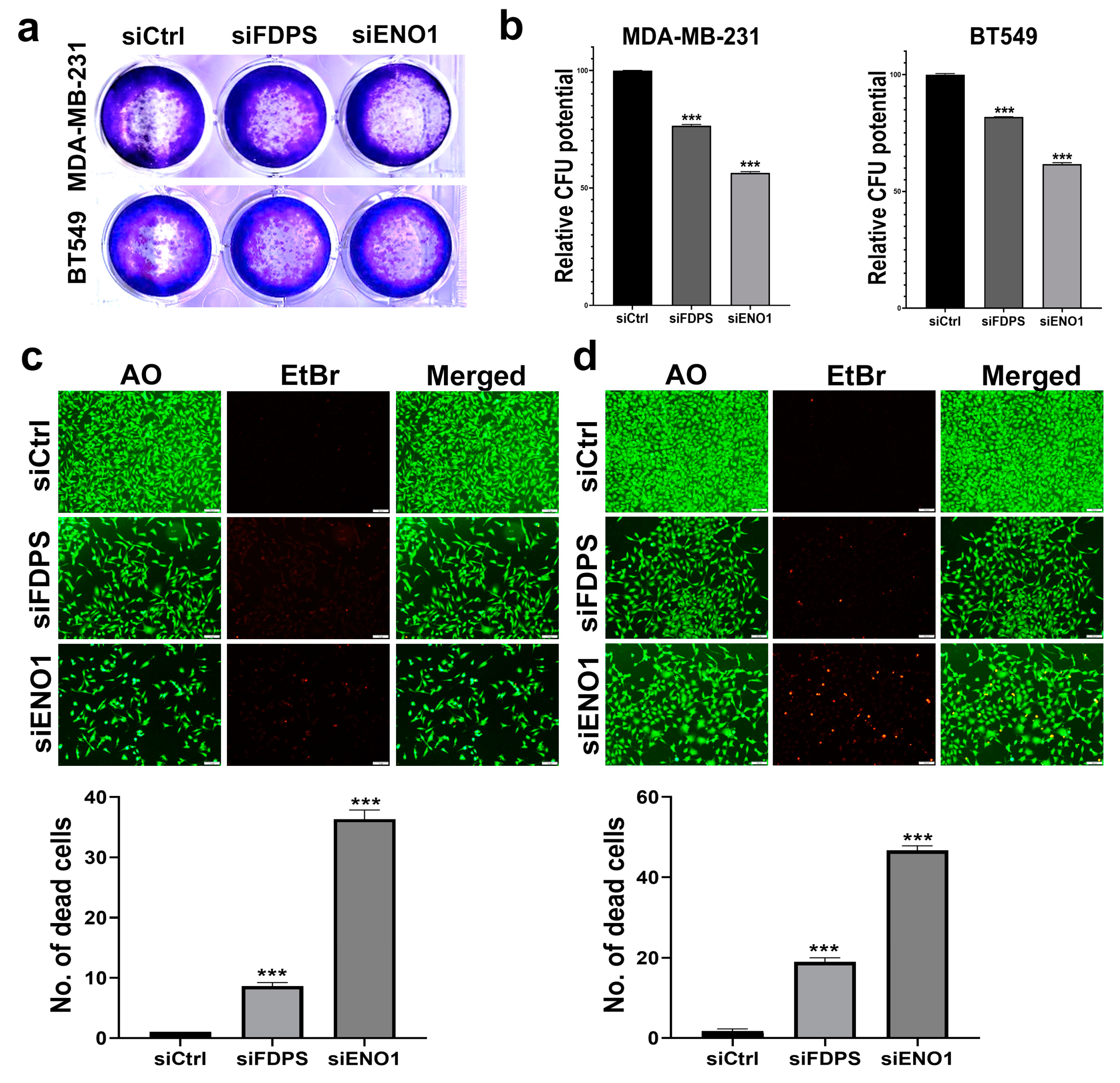
3.5. Targeted Depletion of ENO1 and FDPS Reduces TNBC Colony Formation, Viability, Migration, and Growth under 3D Organotypic Culture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Lukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanislawek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers 2021, 13, 4278. [Google Scholar] [CrossRef]
- Elango, R.; Vishnubalaji, R.; Shaath, H.; Alajez, N.M. Molecular subtyping and functional validation of TTK, TPX2, UBE2C, and LRP8 in sensitivity of TNBC to paclitaxel. Mol. Ther. Methods Clin. Dev. 2021, 20, 601–614. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Venkatasubramanian, M.; Chetal, K.; Schnell, D.J.; Atluri, G.; Salomonis, N. Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF. Bioinformatics 2020, 36, 3773–3780. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Abdel-Razeq, H.; Gehani, S.; Albagha, O.M.E.; Alajez, N.M. Identification of a Gene Panel Predictive of Triple-Negative Breast Cancer Response to Neoadjuvant Chemotherapy Employing Transcriptomic and Functional Validation. Int. J. Mol. Sci. 2022, 23, 10901. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Elango, R.; Alajez, N.M. LncRNA-Based Classification of Triple Negative Breast Cancer Revealed Inherent Tumor Heterogeneity and Vulnerabilities. Noncoding RNA 2022, 8, 44. [Google Scholar] [CrossRef]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Shaath, H.; Elango, R.; Alajez, N.M. Molecular Classification of Breast Cancer Utilizing Long Non-Coding RNA (lncRNA) Transcriptomes Identifies Novel Diagnostic lncRNA Panel for Triple-Negative Breast Cancer. Cancers 2021, 13, 5350. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Elango, R.; Manikandan, M.; Siyal, A.A.; Ali, D.; Al-Rikabi, A.; Hamam, D.; Hamam, R.; Benabdelkamel, H.; Masood, A.; et al. MicroRNA-3148 acts as molecular switch promoting malignant transformation and adipocytic differentiation of immortalized human bone marrow stromal cells via direct targeting of the SMAD2/TGFbeta pathway. Cell Death Discov. 2020, 6, 79. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Januskeviciene, I.; Petrikaite, V. Heterogeneity of breast cancer: The importance of interaction between different tumor cell populations. Life Sci. 2019, 239, 117009. [Google Scholar] [CrossRef]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothe, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Q.; Yu, C.; Cao, Y.; Zuo, Y.; Yang, L. Immune cell infiltration-based signature for prognosis and immunogenomic analysis in breast cancer. Brief Bioinform. 2021, 22, 2020–2031. [Google Scholar] [CrossRef]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Alajez, N.M. Transcriptional landscape associated with TNBC resistance to neoadjuvant chemotherapy revealed by single-cell RNA-seq. Mol. Ther. Oncolytics 2021, 23, 151–162. [Google Scholar] [CrossRef]
- Soto-Perez-de-Celis, E.; Chavarri-Guerra, Y.; Leon-Rodriguez, E.; Gamboa-Dominguez, A. Tumor-Associated Neutrophils in Breast Cancer Subtypes. Asian Pac. J. Cancer Prev. 2017, 18, 2689–2693. [Google Scholar] [CrossRef]
- Liu, F.; Lang, R.; Zhao, J.; Zhang, X.; Pringle, G.A.; Fan, Y.; Yin, D.; Gu, F.; Yao, Z.; Fu, L. CD8+ cytotoxic T cell and FOXP3+ regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res. Treat. 2011, 130, 645–655. [Google Scholar] [CrossRef]
- Li, R.; Jiang, Q.; Tang, C.; Chen, L.; Kong, D.; Zou, C.; Lin, Y.; Luo, J.; Zou, D. Identification of Candidate Genes Associated With Prognosis in Glioblastoma. Front. Mol. Neurosci. 2022, 15, 913328. [Google Scholar] [CrossRef]
- Bi, G.; Zhu, D.; Bian, Y.; Huang, Y.; Zhan, C.; Yang, Y.; Wang, Q. Knockdown of GTF2E2 inhibits the growth and progression of lung adenocarcinoma via RPS4X in vitro and in vivo. Cancer Cell Int. 2021, 21, 181. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, C.; Liu, W.; Chen, W.; Yin, Y.; Li, C.W.; Hsu, J.L.; Sun, J.; Zhou, Q.; Li, H.; et al. SLFN11 inhibits hepatocellular carcinoma tumorigenesis and metastasis by targeting RPS4X via mTOR pathway. Theranostics 2020, 10, 4627–4643. [Google Scholar] [CrossRef]
- Tsofack, S.P.; Meunier, L.; Sanchez, L.; Madore, J.; Provencher, D.; Mes-Masson, A.M.; Lebel, M. Low expression of the X-linked ribosomal protein S4 in human serous epithelial ovarian cancer is associated with a poor prognosis. BMC Cancer 2013, 13, 303. [Google Scholar] [CrossRef]
- Du, C.; Wang, T.; Jia, J.; Li, J.; Xiao, Y.; Wang, J.; Mao, P.; Wang, N.; Shi, L.; Wang, M. Suppression of RPL34 Inhibits Tumor Cell Proliferation and Promotes Apoptosis in Glioblastoma. Appl. Biochem. Biotechnol. 2022, 194, 3494–3506. [Google Scholar] [CrossRef]
- Zhu, Y.; Ren, C.; Jiang, D.; Yang, L.; Chen, Y.; Li, F.; Wang, B.; Zhang, Y. RPL34-AS1-induced RPL34 inhibits cervical cancer cell tumorigenesis via the MDM2-P53 pathway. Cancer Sci. 2021, 112, 1811–1821. [Google Scholar] [CrossRef]
- Liu, H.; Liang, S.; Yang, X.; Ji, Z.; Zhao, W.; Ye, X.; Rui, J. RNAi-mediated RPL34 knockdown suppresses the growth of human gastric cancer cells. Oncol. Rep. 2015, 34, 2267–2272. [Google Scholar] [CrossRef]
- Ropolo, A.; Grasso, D.; Pardo, R.; Sacchetti, M.L.; Archange, C.; Lo Re, A.; Seux, M.; Nowak, J.; Gonzalez, C.D.; Iovanna, J.L.; et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007, 282, 37124–37133. [Google Scholar] [CrossRef]
- Sauermann, M.; Sahin, O.; Sultmann, H.; Hahne, F.; Blaszkiewicz, S.; Majety, M.; Zatloukal, K.; Fuzesi, L.; Poustka, A.; Wiemann, S.; et al. Reduced expression of vacuole membrane protein 1 affects the invasion capacity of tumor cells. Oncogene 2008, 27, 1320–1326. [Google Scholar] [CrossRef]
- Lin, W.; Sun, Y.; Qiu, X.; Huang, Q.; Kong, L.; Lu, J.J. VMP1, a novel prognostic biomarker, contributes to glioma development by regulating autophagy. J. Neuroinflamm. 2021, 18, 165. [Google Scholar] [CrossRef]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non-small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef]
- Yang, Y.; Leonard, M.; Luo, Z.; Yeo, S.; Bick, G.; Hao, M.; Cai, C.; Charif, M.; Wang, J.; Guan, J.L.; et al. Functional cooperation between co-amplified genes promotes aggressive phenotypes of HER2-positive breast cancer. Cell Rep. 2021, 34, 108822. [Google Scholar] [CrossRef]
- Zanin, R.; Pegoraro, S.; Ros, G.; Ciani, Y.; Piazza, S.; Bossi, F.; Bulla, R.; Zennaro, C.; Tonon, F.; Lazarevic, D.; et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J. Exp. Clin. Cancer Res. 2019, 38, 313. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Huang, C.K.; Sun, Y.; Lv, L.; Ping, Y. ENO1 and Cancer. Mol. Ther. Oncolytics 2022, 24, 288–298. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishnubalaji, R.; Alajez, N.M. Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes. Cells 2023, 12, 1182. https://doi.org/10.3390/cells12081182
Vishnubalaji R, Alajez NM. Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes. Cells. 2023; 12(8):1182. https://doi.org/10.3390/cells12081182
Chicago/Turabian StyleVishnubalaji, Radhakrishnan, and Nehad M. Alajez. 2023. "Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes" Cells 12, no. 8: 1182. https://doi.org/10.3390/cells12081182
APA StyleVishnubalaji, R., & Alajez, N. M. (2023). Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes. Cells, 12(8), 1182. https://doi.org/10.3390/cells12081182