

Organoids as an Enabler of Precision Immuno-Oncology


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction and Specifications
2. Approaches to TME Recapitulation or Preservation in Organoids
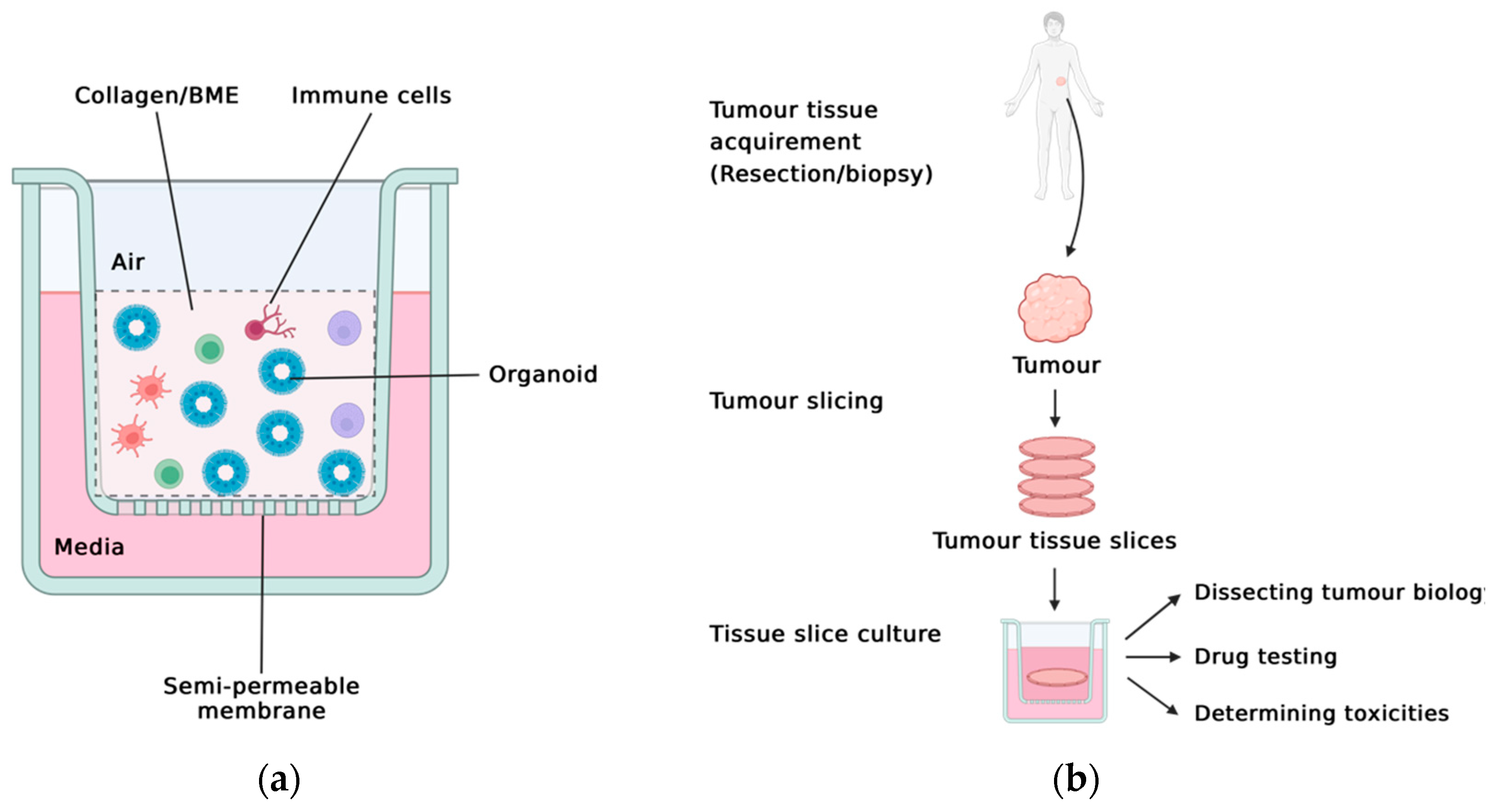
2.1. The Reductionist Approach: Matrigel-Based Organoids
2.2. The Holistic Approach: PDOTS, ALI, TSC
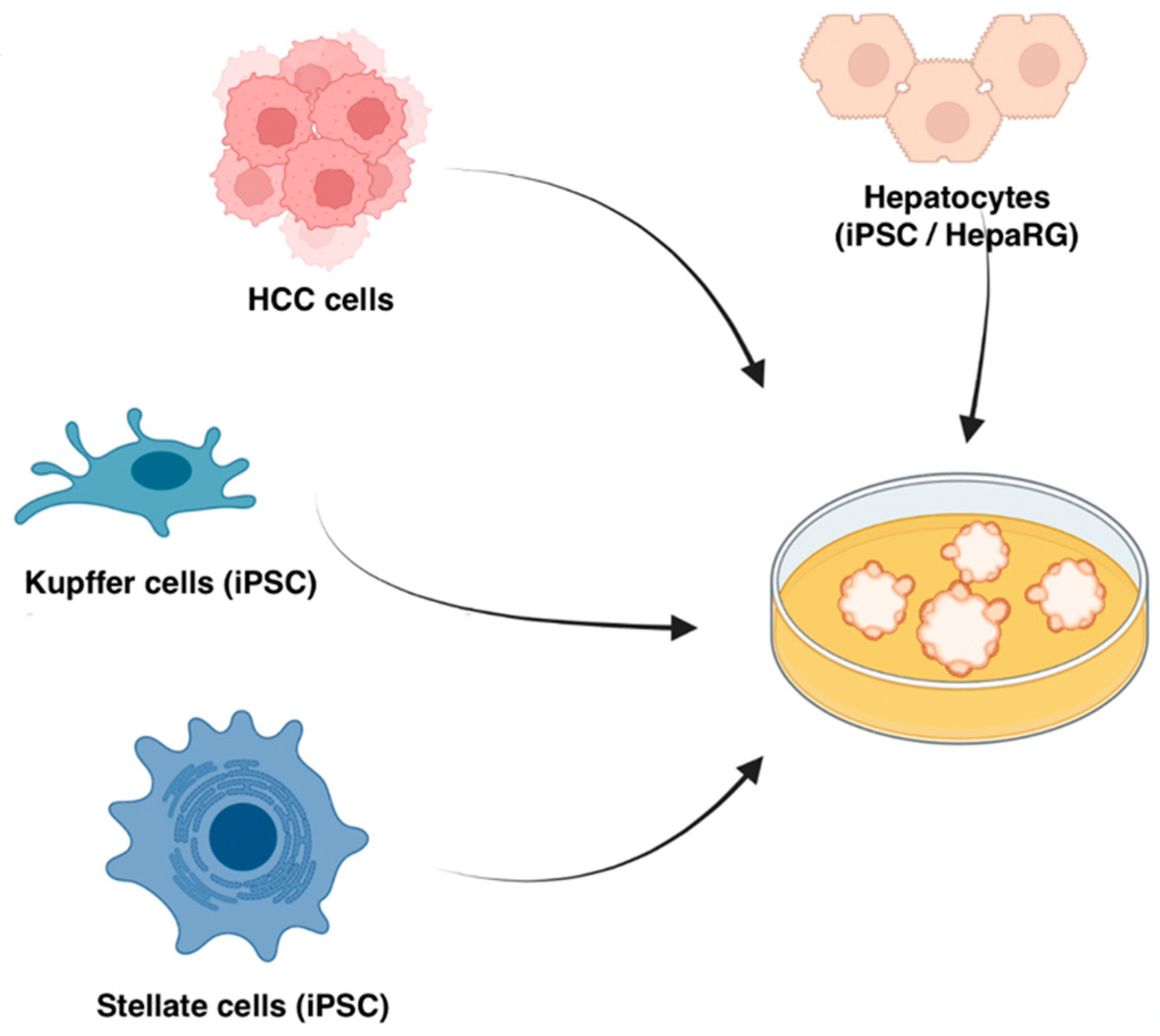
2.3. The Bottom-Up Approach: In Vitro Organoids
3. Translational Immuno-Oncology Research with Organoids
3.1. Investigating T Cell and ICI Responses
3.2. Unravelling the Functions of TME Cells
3.3. Testing of Novel Precision Immuno-Oncology Strategies
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 challenges in cancer immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Orkin, R.W.; Gehron, P.; McGoodwin, E.B.; Martin, G.R.; Valentine, T.; Swarm, R. A murine tumor producing a matrix of basement membrane. J. Exp. Med. 1977, 145, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef]
- Lu, L.G.; Zhou, Z.L.; Wang, X.Y.; Liu, B.Y.; Lu, J.Y.; Liu, S.; Zhang, G.B.; Zhan, M.X.; Chen, Y. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut 2022, 71, 2551–2560. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, 38, e101654. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Clevers, H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014, 24, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.N.; Fong, S.Y.; Tan, W.W.S.; Tan, S.Y.; Liu, M.; Cheng, J.Y.; Lim, S.; Suteja, L.; Huang, E.K.; Chan, J.K.Y.; et al. Establishment and characterization of humanized mouse NPC-PDX model for testing immunotherapy. Cancers 2020, 12, 1025. [Google Scholar] [CrossRef]
- Kang, X.; Kim, J.; Deng, M.; John, S.; Chen, H.; Wu, G.; Phan, H.; Zhang, C.C. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 2016, 15, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; Lopez-Iglesias, C.; Peters, P.J.; Rodriguez-Colman, M.J.; Chuva de Sousa Lopes, S.; et al. Engineered human hepatocyte organoids enable CRISPR-based target discovery and drug screening for steatosis. Nat. Biotechnol. 2023, 1–15. [Google Scholar] [CrossRef]
- Sun, C.P.; Lan, H.R.; Fang, X.L.; Yang, X.Y.; Jin, K.T. Organoid models for precision cancer immunotherapy. Front. Immunol. 2022, 13, 770465. [Google Scholar] [CrossRef]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid models of tumor immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2020, 20, 279–293. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Dao, V.; Yuki, K.; Lo, Y.H.; Nakano, M.; Kuo, C.J. Immune organoids: From tumor modeling to precision oncology. Trends Cancer 2022, 8, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.H.; Karlsson, K.; Kuo, C.J. Applications of organoids for cancer biology and precision medicine. Nat. Cancer 2020, 1, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Gronholm, M.; Feodoroff, M.; Antignani, G.; Martins, B.; Hamdan, F.; Cerullo, V. Patient-derived organoids for precision cancer immunotherapy. Cancer Res. 2021, 81, 3149–3155. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2018, 172, 373–386.e310. [Google Scholar] [CrossRef]
- Lohmussaar, K.; Oka, R.; Espejo Valle-Inclan, J.; Smits, M.H.H.; Wardak, H.; Korving, J.; Begthel, H.; Proost, N.; van de Ven, M.; Kranenburg, O.W.; et al. Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell 2021, 28, 1380–1396. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Boretto, M.; Maenhoudt, N.; Luo, X.; Hennes, A.; Boeckx, B.; Bui, B.; Heremans, R.; Perneel, L.; Kobayashi, H.; Van Zundert, I.; et al. Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol. 2019, 21, 1041–1051. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 2018, 23, 882–897.e811. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 2020, 180, 188–204. [Google Scholar] [CrossRef]
- Tanaka, N.; Osman, A.A.; Takahashi, Y.; Lindemann, A.; Patel, A.A.; Zhao, M.; Takahashi, H.; Myers, J.N. Head and neck cancer organoids established by modification of the CTOS method can be used to predict in vivo drug sensitivity. Oral Oncol. 2018, 87, 49–57. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Knutsdottir, H.; Hui, K.; Weiss, M.J.; He, J.; Philosophe, B.; Cameron, A.M.; Wolfgang, C.L.; Pawlik, T.M.; Ghiaur, G.; et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 2019, 4, e121490. [Google Scholar] [CrossRef] [PubMed]
- Calandrini, C.; Schutgens, F.; Oka, R.; Margaritis, T.; Candelli, T.; Mathijsen, L.; Ammerlaan, C.; van Ineveld, R.L.; Derakhshan, S.; de Haan, S.; et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 2020, 11, 1310. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9, 2983. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lohmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Ogawa, H.; Yoshida, K.; Matsuoka, Y.; Shizuma, K.; Imura, Y.; Tamiya, H.; Nakai, S.; Yagi, T.; Nagata, S.; et al. Establishment of organoids from human epithelioid sarcoma with the air-liquid interface organoid cultures. Front. Oncol. 2022, 12, 893592. [Google Scholar] [CrossRef]
- Mullenders, J.; de Jongh, E.; Brousali, A.; Roosen, M.; Blom, J.P.A.; Begthel, H.; Korving, J.; Jonges, T.; Kranenburg, O.; Meijer, R.; et al. Mouse and human urothelial cancer organoids: A tool for bladder cancer research. Proc. Natl. Acad. Sci. USA 2019, 116, 4567–4574. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Boretto, M.; Millen, R.; Natesh, N.; Reckzeh, E.S.; Hsu, C.; Negrete, M.; Yao, H.; Quayle, W.; Heaton, B.E.; et al. Rapid tissue prototyping with micro-organospheres. Stem Cell Rep. 2022, 17, 1959–1975. [Google Scholar] [CrossRef]
- Ding, S.; Hsu, C.; Wang, Z.; Natesh, N.R.; Millen, R.; Negrete, M.; Giroux, N.; Rivera, G.O.; Dohlman, A.; Bose, S.; et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell 2022, 29, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid modeling of the tumor immune microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef]
- Jabbari, N.; Kenerson, H.L.; Lausted, C.; Yan, X.; Meng, C.; Sullivan, K.M.; Baloni, P.; Bergey, D.; Pillarisetty, V.G.; Hood, L.E.; et al. Modulation of immune checkpoints by chemotherapy in human colorectal liver metastases. Cell Rep. Med. 2020, 1, 100160. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef]
- Mathis, C.; Poussin, C.; Weisensee, D.; Gebel, S.; Hengstermann, A.; Sewer, A.; Belcastro, V.; Xiang, Y.; Ansari, S.; Wagner, S.; et al. Human bronchial epithelial cells exposed in vitro to cigarette smoke at the air-liquid interface resemble bronchial epithelium from human smokers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L489–L503. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Starner, T.D.; Scheetz, T.E.; Traver, G.L.; Tilley, A.E.; Harvey, B.G.; Crystal, R.G.; McCray, P.B., Jr.; Zabner, J. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L25–L31. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Choudhury, D.; Fang, Y.; Hunziker, W. Towards manufacturing of human organoids. Biotechnol. Adv. 2020, 39, 107460. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Luo, C.; Li, C.; Huang, J.; Liu, F. Organoids to study immune functions, immunological diseases and immunotherapy. Cancer Lett. 2020, 477, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G.; Yeung, R.S. Protocol for tissue slice cultures from human solid tumors to study therapeutic response. STAR Protoc. 2021, 2, 100574. [Google Scholar] [CrossRef]
- Chadwick, E.J.; Yang, D.P.; Filbin, M.G.; Mazzola, E.; Sun, Y.; Behar, O.; Pazyra-Murphy, M.F.; Goumnerova, L.; Ligon, K.L.; Stiles, C.D.; et al. A brain tumor/organotypic slice co-culture system for studying tumor microenvironment and targeted drug therapies. J. Vis. Exp. 2015, 105, e53304. [Google Scholar] [CrossRef]
- Naipal, K.A.; Verkaik, N.S.; Sanchez, H.; van Deurzen, C.H.; den Bakker, M.A.; Hoeijmakers, J.H.; Kanaar, R.; Vreeswijk, M.P.; Jager, A.; van Gent, D.C. Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer 2016, 16, 78. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef]
- Voabil, P.; de Bruijn, M.; Roelofsen, L.M.; Hendriks, S.H.; Brokamp, S.; van den Braber, M.; Broeks, A.; Sanders, J.; Herzig, P.; Zippelius, A.; et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 2021, 27, 1250–1261. [Google Scholar] [CrossRef]
- Karekla, E.; Liao, W.J.; Sharp, B.; Pugh, J.; Reid, H.; Quesne, J.L.; Moore, D.; Pritchard, C.; MacFarlane, M.; Pringle, J.H. Ex vivo explant cultures of non-small cell lung carcinoma enable evaluation of primary tumor responses to anticancer therapy. Cancer Res. 2017, 77, 2029–2039. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Sullivan, K.M.; Seo, Y.D.; Stadeli, K.M.; Ussakli, C.; Yan, X.; Lausted, C.; Pillarisetty, V.G.; Park, J.O.; Riehle, K.J.; et al. Tumor slice culture as a biologic surrogate of human cancer. Ann. Transl. Med. 2020, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Moro, C.F.; Del Chiaro, M.; Pouso, S.; Sebestyen, A.; Lohr, M.; Bjornstedt, M.; Verbeke, C.S. Ex vivo organotypic culture system of precision-cut slices of human pancreatic ductal adenocarcinoma. Sci. Rep. 2019, 9, 2133. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Schiewer, M.J.; de Leeuw, R.; Dylgjeri, E.; McCue, P.A.; Shah, N.; Gomella, L.G.; Lallas, C.D.; Trabulsi, E.J.; Centenera, M.M.; et al. Patient-derived models reveal impact of the tumor microenvironment on therapeutic response. Eur. Urol. Oncol. 2018, 1, 325–337. [Google Scholar] [CrossRef] [PubMed]
- van de Merbel, A.F.; van der Horst, G.; van der Mark, M.H.; van Uhm, J.I.M.; van Gennep, E.J.; Kloen, P.; Beimers, L.; Pelger, R.C.M.; van der Pluijm, G. An ex vivo tissue culture model for the assessment of individualized drug responses in prostate and bladder cancer. Front. Oncol. 2018, 8, 400. [Google Scholar] [CrossRef]
- Majumder, B.; Baraneedharan, U.; Thiyagarajan, S.; Radhakrishnan, P.; Narasimhan, H.; Dhandapani, M.; Brijwani, N.; Pinto, D.D.; Prasath, A.; Shanthappa, B.U.; et al. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat. Commun. 2015, 6, 6169. [Google Scholar] [CrossRef]
- Sonnichsen, R.; Hennig, L.; Blaschke, V.; Winter, K.; Korfer, J.; Hahnel, S.; Monecke, A.; Wittekind, C.; Jansen-Winkeln, B.; Thieme, R.; et al. Individual susceptibility analysis using patient-derived slice cultures of colorectal carcinoma. Clin. Colorectal. Cancer 2018, 17, e189–e199. [Google Scholar] [CrossRef]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Cen, J.; Ma, X.; Cui, L.; Qiu, Z.; Zhang, Z.; Li, H.; Yang, R.Z.; Wang, C.; et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 2019, 21, 1015–1026. [Google Scholar] [CrossRef]
- Cai, J.; Zhao, Y.; Liu, Y.; Ye, F.; Song, Z.; Qin, H.; Meng, S.; Chen, Y.; Zhou, R.; Song, X.; et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology 2007, 45, 1229–1239. [Google Scholar] [CrossRef]
- Tasnim, F.; Xing, J.; Huang, X.; Mo, S.; Wei, X.; Tan, M.H.; Yu, H. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials 2019, 192, 377–391. [Google Scholar] [CrossRef]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdu, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of hepatic stellate cells from human pluripotent stem cells enables in vitro modeling of liver fibrosis. Cell Stem Cell 2018, 23, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Enomura, M.; Yoshizawa, E.; Kimura, M.; Koike, H.; Ueno, Y.; Matsuzaki, T.; Yamazaki, T.; Toyohara, T.; Osafune, K.; et al. Vascularized and complex organ buds from diverse tissues via mesenchymal cell-driven condensation. Cell Stem Cell 2015, 16, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by mesenchymal stem cells (MSCs): Mechanisms of action of living, apoptotic, and dead mscs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab. 2019, 30, 374–384.e376. [Google Scholar] [CrossRef]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.R.; Dunn, A.; et al. High-fidelity drug-induced liver injury screen using human pluripotent stem cell-derived organoids. Gastroenterology 2021, 160, 831–846.e810. [Google Scholar] [CrossRef]
- Khan, A.O.; Rodriguez-Romera, A.; Reyat, J.S.; Olijnik, A.A.; Colombo, M.; Wang, G.; Wen, W.X.; Sousos, N.; Murphy, L.C.; Grygielska, B.; et al. Human bone marrow organoids for disease modelling, discovery and validation of therapeutic targets in hematological malignancies. Cancer Discov. 2023, 13, 364–385. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018, 174, 1586–1598.e1512. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid-T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Alieva, M.; Cleven, A.; Keramati, F.; Wezenaar, A.K.L.; van Vliet, E.J.; Puschhof, J.; Brazda, P.; Johanna, I.; Meringa, A.D.; et al. Uncovering the mode of action of engineered T cells in patient cancer organoids. Nat. Biotechnol. 2022, 41, 60–69. [Google Scholar] [CrossRef]
- Forsythe, S.D.; Erali, R.A.; Sasikumar, S.; Laney, P.; Shelkey, E.; D’Agostino, R., Jr.; Miller, L.D.; Shen, P.; Levine, E.A.; Soker, S.; et al. Organoid platform in preclinical investigation of personalized immunotherapy efficacy in appendiceal cancer: Feasibility study. Clin. Cancer Res. 2021, 27, 5141–5150. [Google Scholar] [CrossRef]
- Gao, Y.; Bi, D.; Xie, R.; Li, M.; Guo, J.; Liu, H.; Guo, X.; Fang, J.; Ding, T.; Zhu, H.; et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 398. [Google Scholar] [CrossRef]
- Scanu, T.; Spaapen, R.M.; Bakker, J.M.; Pratap, C.B.; Wu, L.E.; Hofland, I.; Broeks, A.; Shukla, V.K.; Kumar, M.; Janssen, H.; et al. Salmonella manipulation of host signaling pathways provokes cellular transformation associated with gallbladder carcinoma. Cell Host Microbe. 2015, 17, 763–774. [Google Scholar] [CrossRef]
- McCracken, K.W.; Cata, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef]
- Xu, H.; Van der Jeught, K.; Zhou, Z.; Zhang, L.; Yu, T.; Sun, Y.; Li, Y.; Wan, C.; So, K.M.; Liu, D.; et al. Atractylenolide I enhances responsiveness to immune checkpoint blockade therapy by activating tumor antigen presentation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Zhou, Z.; Van der Jeught, K.; Fang, Y.; Yu, T.; Li, Y.; Ao, Z.; Liu, S.; Zhang, L.; Yang, Y.; Eyvani, H.; et al. An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat. Biomed. Eng. 2021, 5, 1320–1335. [Google Scholar] [CrossRef]
- Scognamiglio, G.; De Chiara, A.; Parafioriti, A.; Armiraglio, E.; Fazioli, F.; Gallo, M.; Aversa, L.; Camerlingo, R.; Cacciatore, F.; Colella, G.; et al. Patient-derived organoids as a potential model to predict response to PD-1/PD-L1 checkpoint inhibitors. Br. J. Cancer 2019, 121, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Gordon, S.; Imhof, B.A.; Nunez, G.; Bousso, P. Elie Metchnikoff (1845–1916): Celebrating 100 years of cellular immunology and beyond. Nat. Rev. Immunol. 2016, 16, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Yeku, O.O.; Rafiq, S.; Purdon, T.; Dong, X.; Zhu, L.; Zhang, T.; Wang, H.; Yu, Z.; Mai, J.; et al. Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages. Nat. Commun. 2020, 11, 6298. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Smith, S.E.; Chen, S.; Li, H.; Wu, D.; Meneses-Giles, P.I.; Wang, Y.; Hembree, M.; Yi, K.; Zhao, X.; et al. Tumor-initiating stem cell shapes its microenvironment into an immunosuppressive barrier and pro-tumorigenic niche. Cell Rep. 2021, 36, 109674. [Google Scholar] [CrossRef]
- Haque, M.R.; Wessel, C.R.; Leary, D.D.; Wang, C.; Bhushan, A.; Bishehsari, F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst. Nanoeng. 2022, 8, 36. [Google Scholar] [CrossRef]
- Fang, H.; Huang, Y.; Luo, Y.; Tang, J.; Yu, M.; Zhang, Y.; Zhong, M. SIRT1 induces the accumulation of TAMs at colorectal cancer tumor sites via the CXCR4/CXCL12 axis. Cell Immunol. 2022, 371, 104458. [Google Scholar] [CrossRef]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Lim, J.T.C.; Kwang, L.G.; Ho, N.C.W.; Toh, C.C.M.; Too, N.S.H.; Hooi, L.; Benoukraf, T.; Chow, P.K.; Dan, Y.Y.; Chow, E.K.; et al. Hepatocellular carcinoma organoid co-cultures mimic angiocrine crosstalk to generate inflammatory tumor microenvironment. Biomaterials 2022, 284, 121527. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Morales, R.T.; Qian, W.; Wang, H.; Gagner, J.P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Koh, V.; Chakrabarti, J.; Torvund, M.; Steele, N.; Hawkins, J.A.; Ito, Y.; Wang, J.; Helmrath, M.A.; Merchant, J.L.; Ahmed, S.A.; et al. Hedgehog transcriptional effector GLI mediates mTOR-Induced PD-L1 expression in gastric cancer organoids. Cancer Lett. 2021, 518, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and human-derived autologous organoid/immune cell co-cultures as pre-clinical models of pancreatic ductal adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Rong, Y.; Tang, X.; Yi, K.; Qi, P.; Hou, J.; Liu, W.; He, Y.; Gao, X.; Yuan, C.; et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer 2022, 21, 45. [Google Scholar] [CrossRef]
- Cao, K.; Zhang, G.; Zhang, X.; Yang, M.; Wang, Y.; He, M.; Lu, J.; Liu, H. Stromal infiltrating mast cells identify immunoevasive subtype high-grade serous ovarian cancer with poor prognosis and inferior immunotherapeutic response. Oncoimmunology 2021, 10, 1969075. [Google Scholar] [CrossRef]
- Frede, A.; Czarnewski, P.; Monasterio, G.; Tripathi, K.P.; Bejarano, D.A.; Ramirez Flores, R.O.; Sorini, C.; Larsson, L.; Luo, X.; Geerlings, L.; et al. B cell expansion hinders the stroma-epithelium regenerative cross talk during mucosal healing. Immunity 2022, 55, 2336–2351. [Google Scholar] [CrossRef]
- Purwada, A.; Singh, A. Immuno-engineered organoids for regulating the kinetics of B-cell development and antibody production. Nat. Protoc. 2017, 12, 168–182. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, V.L.; Henriet, E.; Linville, R.M.; Wong, A.D.; Searson, P.C.; Ewald, A.J. A tissue-engineered 3D microvessel model reveals the dynamics of mosaic vessel formation in breast cancer. Cancer Res. 2020, 80, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Mazio, C.; Casale, C.; Imparato, G.; Urciuolo, F.; Netti, P.A. Recapitulating spatiotemporal tumor heterogeneity in vitro through engineered breast cancer microtissues. Acta Biomater. 2018, 73, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity 2019, 51, 655–670.e658. [Google Scholar] [CrossRef]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.A.; Pai, R.; Bleriot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 2020, 183, 377–394.e321. [Google Scholar] [CrossRef]
- Winkler, M.; Staniczek, T.; Kurschner, S.W.; Schmid, C.D.; Schonhaber, H.; Cordero, J.; Kessler, L.; Mathes, A.; Sticht, C.; Nessling, M.; et al. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J. Hepatol. 2021, 74, 380–393. [Google Scholar] [CrossRef]
- Choi, J.I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53. [Google Scholar] [CrossRef]
- Lai Benjamin, F.L.; Lu Rick, X.; Hu, Y.; Davenport, H.L.; Dou, W.; Wang, E.Y.; Radulovich, N.; Tsao, M.S.; Sun, Y.; Radisic, M. Recapitulating pancreatic tumor microenvironment through synergistic use of patient organoids and organ-on-a-chip vasculature. Adv. Funct. Mater. 2020, 30, 2000545. [Google Scholar] [CrossRef]
- Hoarau-Vechot, J.; Blot-Dupin, M.; Pauly, L.; Touboul, C.; Rafii, S.; Rafii, A.; Pasquier, J. Akt-activated endothelium increases cancer cell proliferation and resistance to treatment in ovarian cancer cell organoids. Int. J. Mol. Sci. 2022, 23, 14173. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wan, Z.; Kamm, R.D. Vascularized organoids on a chip: Strategies for engineering organoids with functional vasculature. Lab. Chip. 2021, 21, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology 2020, 71, 1247–1261. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor endothelial heterogeneity in cancer progression. Cancers 2019, 11, 1511. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Grunwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e5518. [Google Scholar] [CrossRef]
- Luo, X.; Fong, E.L.S.; Zhu, C.; Lin, Q.X.X.; Xiong, M.; Li, A.; Li, T.; Benoukraf, T.; Yu, H.; Liu, S. Hydrogel-based colorectal cancer organoid co-culture models. Acta. Biomater. 2021, 132, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, V.S.; de Jesus Cardona, C.; Hejret, V.; Tiefenbacher, A.; Mair, T.; Tran, L.; Pfneissl, J.; Draganić, K.; Binder, C.; Kabiljo, J.; et al. Mimicking tumor cell heterogeneity of colorectal cancer in a patient-derived organoid-fibroblast model. Cell. Mol. Gastroenterol. Hepatol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.A.; Ma, B.; Su, J.; et al. Cancer-associated fibroblasts provide a stromal niche for liver cancer organoids that confers trophic effects and therapy resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor microenvironment-derived NRG1 promotes antiandrogen resistance in prostate cancer. Cancer Cell 2020, 38, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Ooft, S.N.; Weeber, F.; Schipper, L.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van de Haar, J.; Prevoo, W.; van Werkhoven, E.; Snaebjornsson, P.; et al. Prospective experimental treatment of colorectal cancer patients based on organoid drug responses. ESMO Open 2021, 6, 100103. [Google Scholar] [CrossRef] [PubMed]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef]
- Gutting, T.; Hauber, V.; Pahl, J.; Klapproth, K.; Wu, W.; Dobrota, I.; Herweck, F.; Reichling, J.; Helm, L.; Schroeder, T.; et al. PPARgamma induces PD-L1 expression in MSS+ colorectal cancer cells. Oncoimmunology 2021, 10, 1906500. [Google Scholar] [CrossRef]
- Sun, Y.; Revach, O.Y.; Anderson, S.; Kessler, E.A.; Wolfe, C.H.; Jenney, A.; Mills, C.E.; Robitschek, E.J.; Davis, T.G.R.; Kim, S.; et al. Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature 2023, 615, 158–167. [Google Scholar] [CrossRef]
- Kirchhammer, N.; Trefny, M.P.; Auf der Maur, P.; Laubli, H.; Zippelius, A. Combination cancer immunotherapies: Emerging treatment strategies adapted to the tumor microenvironment. Sci. Transl. Med. 2022, 14, eabo3605. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid cell-derived arginase in cancer immune response. Front Immunol. 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dong, J.; Zheng, Y.; Zhou, J.; Yuan, Y.; Ta, H.M.; Miller, H.E.; Olson, M.; Rajasekaran, K.; Ernstoff, M.S.; et al. Immune-checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell-mediated inflammation and immunosuppression. Cancer Immunol. Res. 2019, 7, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Mo, S.; Sun, Z.; Lu, Z.; Yu, S.; Chen, J.; Xiang, Y. Analysis of the immune checkpoint V-domain Ig-containing suppressor of T-cell activation (VISTA) in endometrial cancer. Mod. Pathol. 2022, 35, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, D.; Paliwal, S.; Dharmadhikari, B.; Guan, S.; Liu, L.; Kar, S.; Tulsian, N.K.; Gruber, J.J.; DiMascio, L.; Paszkiewicz, K.H.; et al. Rationally targeted anti-VISTA antibody that blockades the C-C’ loop region can reverse VISTA immune suppression and remodel the immune microenvironment to potently inhibit tumor growth in an Fc independent manner. J. Immunother. Cancer 2022, 10, e003382. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Y.; Li, Q.; Yao, J.; Yuan, X.; Zhang, Y.; Yin, X.; Saito, Y.; Fan, H.; Li, P.; et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients via activation of the macrophage histamine receptor H1. Cancer Cell 2022, 40, 36–52.e39. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.H.; He, A.R.; Ryoo, B.Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gieniec, K.A.; Wright, J.A.; Wang, T.; Asai, N.; Mizutani, Y.; Lida, T.; Ando, R.; Suzuki, N.; Lannagan, T.R.M.; et al. The balance of stromal BMP signaling mediated by GREM1 and ISLR drives colorectal carcinogenesis. Gastroenterology 2021, 160, 1224–1239.e1230. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef]
- Lowery, F.J.; Krishna, S.; Yossef, R.; Parikh, N.B.; Chatani, P.D.; Zacharakis, N.; Parkhurst, M.R.; Levin, N.; Sindiri, S.; Sachs, A.; et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 2022, 375, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yuan, T.; Ma, L.; Zhu, Y.; Bao, J.; Zhao, X.; Zhao, Y.; Zong, Y.; Zhang, Y.; Yang, S.; et al. Hepatobiliary tumor organoids reveal HLA class I neoantigen landscape and antitumoral activity of neoantigen peptide enhanced with immune checkpoint inhibitors. Adv. Sci. 2022, 9, e2105810. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Sacks, N.J.; Schenkel, J.M.; Ely, Z.A.; Smith, O.; Hauck, H.; Jaeger, A.M.; Zhang, D.; Backlund, C.M.; Beytagh, M.C.; et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat. Cancer 2021, 2, 1071–1085. [Google Scholar] [CrossRef]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Demmers, L.C.; Kretzschmar, K.; Van Hoeck, A.; Bar-Epraim, Y.E.; van den Toorn, H.W.P.; Koomen, M.; van Son, G.; van Gorp, J.; Pronk, A.; Smakman, N.; et al. Single-cell derived tumor organoids display diversity in HLA class I peptide presentation. Nat. Commun. 2020, 11, 5338. [Google Scholar] [CrossRef]
- Liu, T.; Tan, J.; Wu, M.; Fan, W.; Wei, J.; Zhu, B.; Guo, J.; Wang, S.; Zhou, P.; Zhang, H.; et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39(+)CD8(+) T cells. Gut 2021, 70, 1965–1977. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Tan, J.; Liu, T.; Liu, B.; Tang, Y.; Zhang, H.; Li, J. The CD39(+) HBV surface protein-targeted CAR-T and personalized tumor-reactive CD8(+) T cells exhibit potent anti-HCC activity. Mol. Ther. 2021, 29, 1794–1807. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, H.; Yu, L.; Wang, J.; Meng, Q.; Mei, H.; Cai, Z.; Chen, W.; Huang, W. Patient-derived renal cell carcinoma organoids for personalized cancer therapy. Clin. Transl. Med. 2022, 12, e970. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. Gammadelta T cells: Pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef]
- Wan, C.; Keany, M.P.; Dong, H.; Al-Alem, L.F.; Pandya, U.M.; Lazo, S.; Boehnke, K.; Lynch, K.N.; Xu, R.; Zarrella, D.T.; et al. Enhanced efficacy of simultaneous PD-1 and PD-L1 immune checkpoint blockade in high-grade serous ovarian cancer. Cancer Res. 2021, 81, 158–173. [Google Scholar] [CrossRef]
- de Vries, N.L.; van de Haar, J.; Veninga, V.; Chalabi, M.; Ijsselsteijn, M.E.; van der Ploeg, M.; van den Bulk, J.; Ruano, D.; van den Berg, J.G.; Haanen, J.B.; et al. Gammadelta T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature 2023, 613, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Wang, H.; Liu, Q.; Zhang, J.; Lu, H.; Luo, L.; Shi, C.; Lin, S.; Dong, L.; Guo, Y.; et al. Dichotomous and stable gamma delta T-cell number and function in healthy individuals. J. Immunother. Cancer 2021, 9, e002274. [Google Scholar] [CrossRef] [PubMed]
- Knelson, E.H.; Ivanova, E.V.; Tarannum, M.; Campisi, M.; Lizotte, P.H.; Booker, M.A.; Ozgenc, I.; Noureddine, M.; Meisenheimer, B.; Chen, M.; et al. Activation of tumor-cell STING primes NK-cell therapy. Cancer Immunol. Res. 2022, 10, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Watts, G. Jacques Miller: Immunologist who discovered role of the thymus. Lancet 2011, 378, 1290. [Google Scholar] [CrossRef]
- Yanagi, Y.; Yoshikai, Y.; Leggett, K.; Clark, S.P.; Aleksander, I.; Mak, T.W. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature 1984, 308, 145–149. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Ernberg, I.; Karre, K.; Wigzell, H. George Klein (1925–2016). Nature 2017, 542, 296. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Wadman, M. FDA no longer has to require animal testing for new drugs. Science 2023, 379, 127–128. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Fong, A.; Seow, S.V.; Toh, H.C. Organoids as an Enabler of Precision Immuno-Oncology. Cells 2023, 12, 1165. https://doi.org/10.3390/cells12081165
Zhao J, Fong A, Seow SV, Toh HC. Organoids as an Enabler of Precision Immuno-Oncology. Cells. 2023; 12(8):1165. https://doi.org/10.3390/cells12081165
Chicago/Turabian StyleZhao, Junzhe, Antoinette Fong, See Voon Seow, and Han Chong Toh. 2023. "Organoids as an Enabler of Precision Immuno-Oncology" Cells 12, no. 8: 1165. https://doi.org/10.3390/cells12081165
APA StyleZhao, J., Fong, A., Seow, S. V., & Toh, H. C. (2023). Organoids as an Enabler of Precision Immuno-Oncology. Cells, 12(8), 1165. https://doi.org/10.3390/cells12081165