
Molecular Spectroscopy Evidence of 1,3,5-Tris(4-carboxyphenyl)benzene Binding to DNA: Anticancer Potential along with the Comparative Binding Profile of Intercalation via Modeling Studies


Abstract
1. Introduction
2. Experimental
2.1. Cell Viability Assay
2.2. Spectrophotometric DNA Binding Analysis
2.3. Computational Investigations
2.3.1. Density Functional Theory Calculations
2.3.2. Molecular Docking Studies
2.4. Molecular Dynamics Simulations
3. Result and Discussions
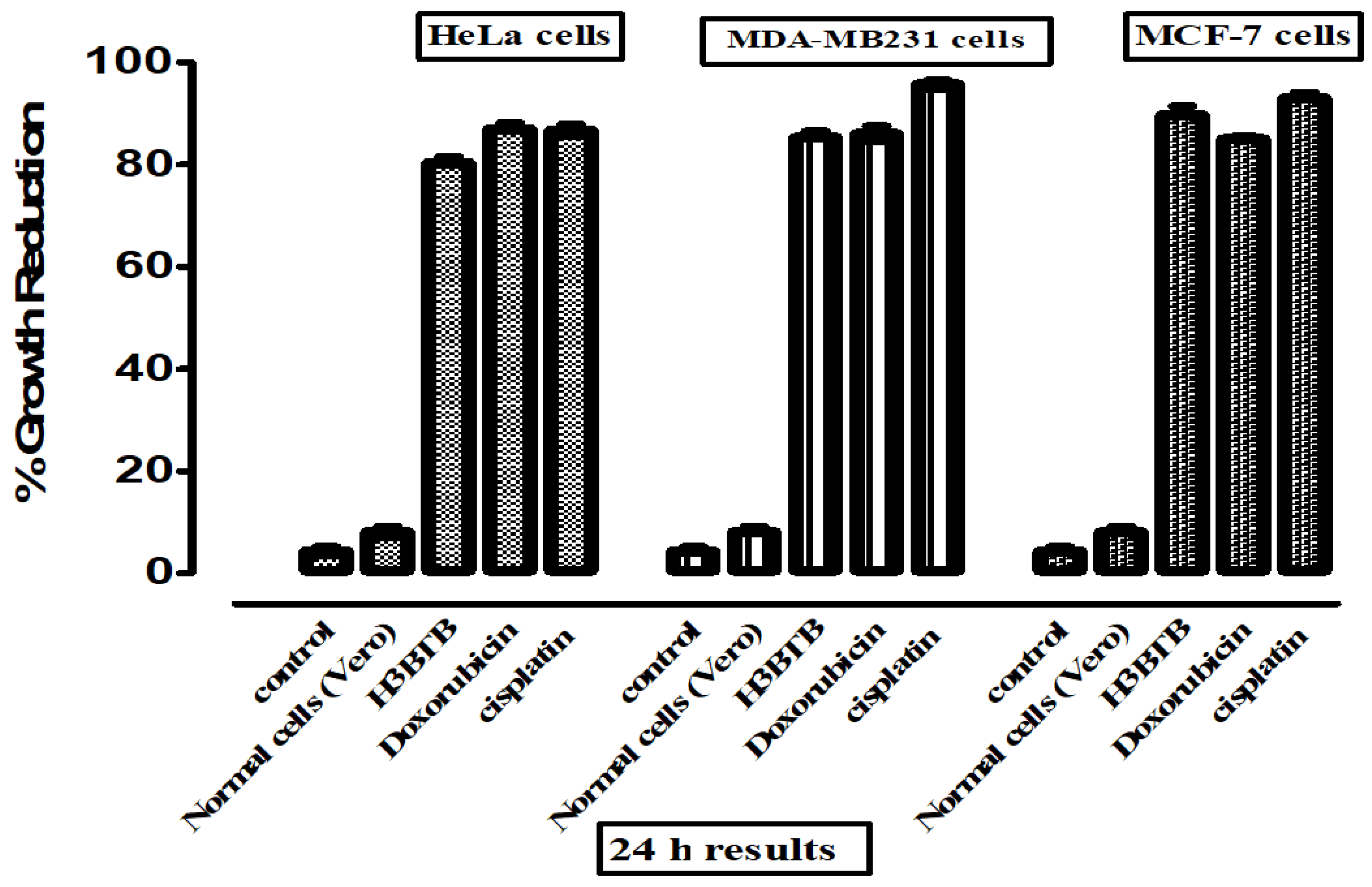
3.1. In Vitro Cytotoxic Activity
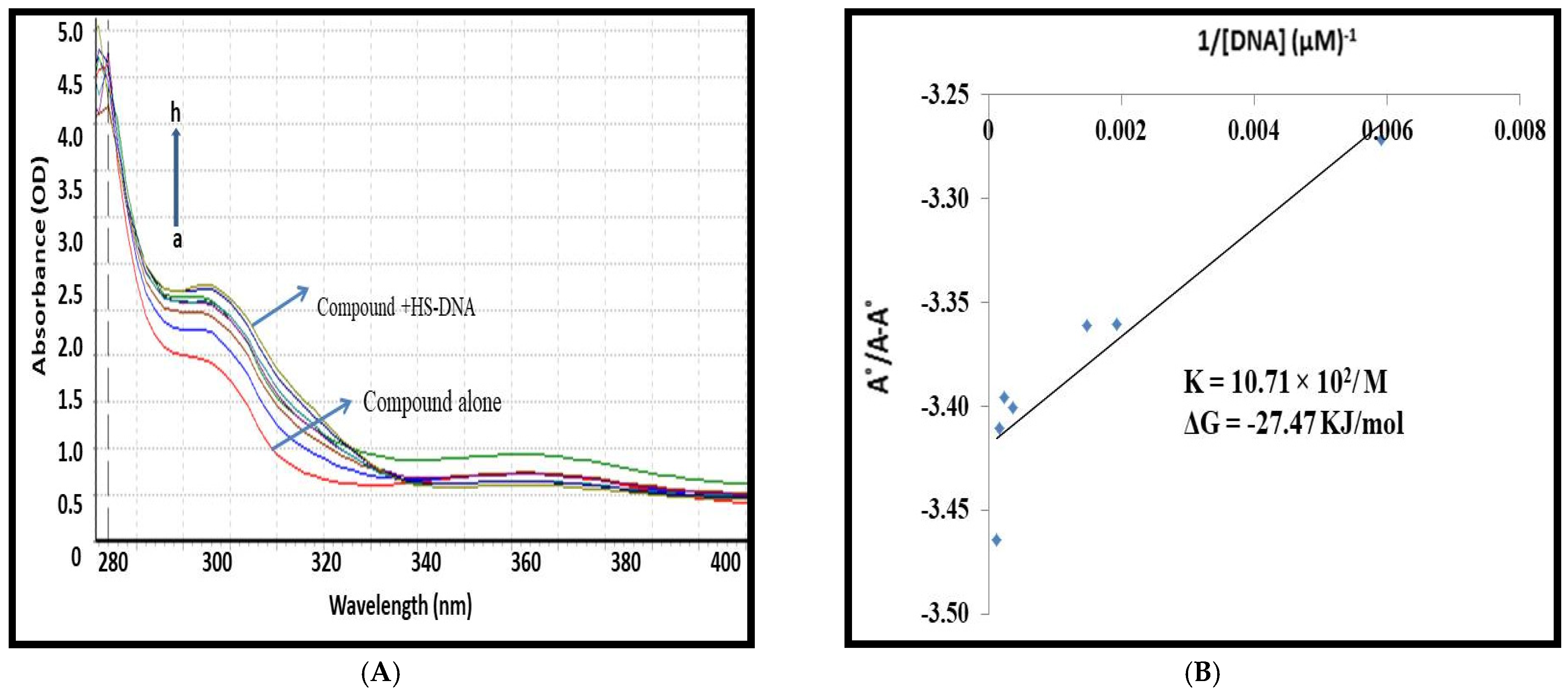
3.2. Spectroscopic Studies for 1,3,5-Tris(4-carboxyphenyl)benzene–DNA Binding
3.3. Density Functional Theory Calculations
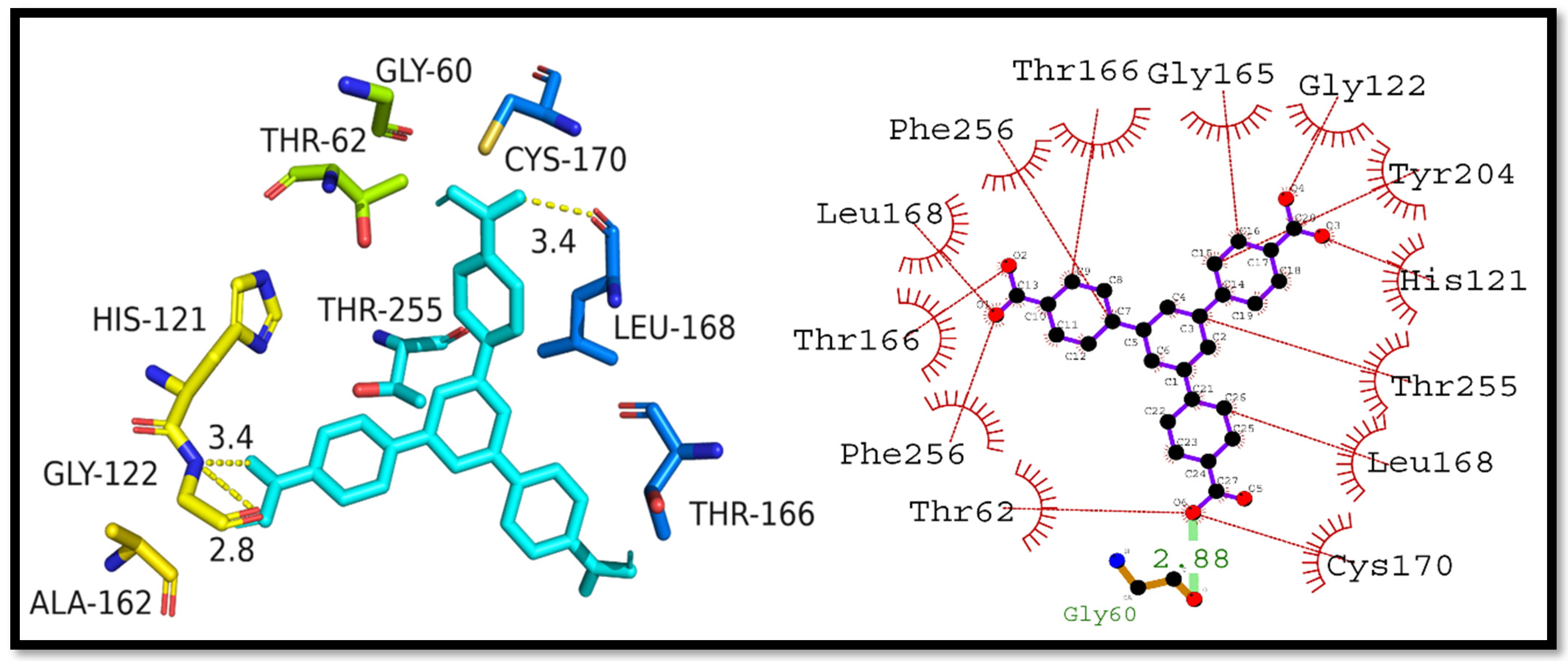
3.4. Molecular Docking Studies
3.5. Interpretation of Molecular Interactions
3.6. Intercalation of Nucleic Acids
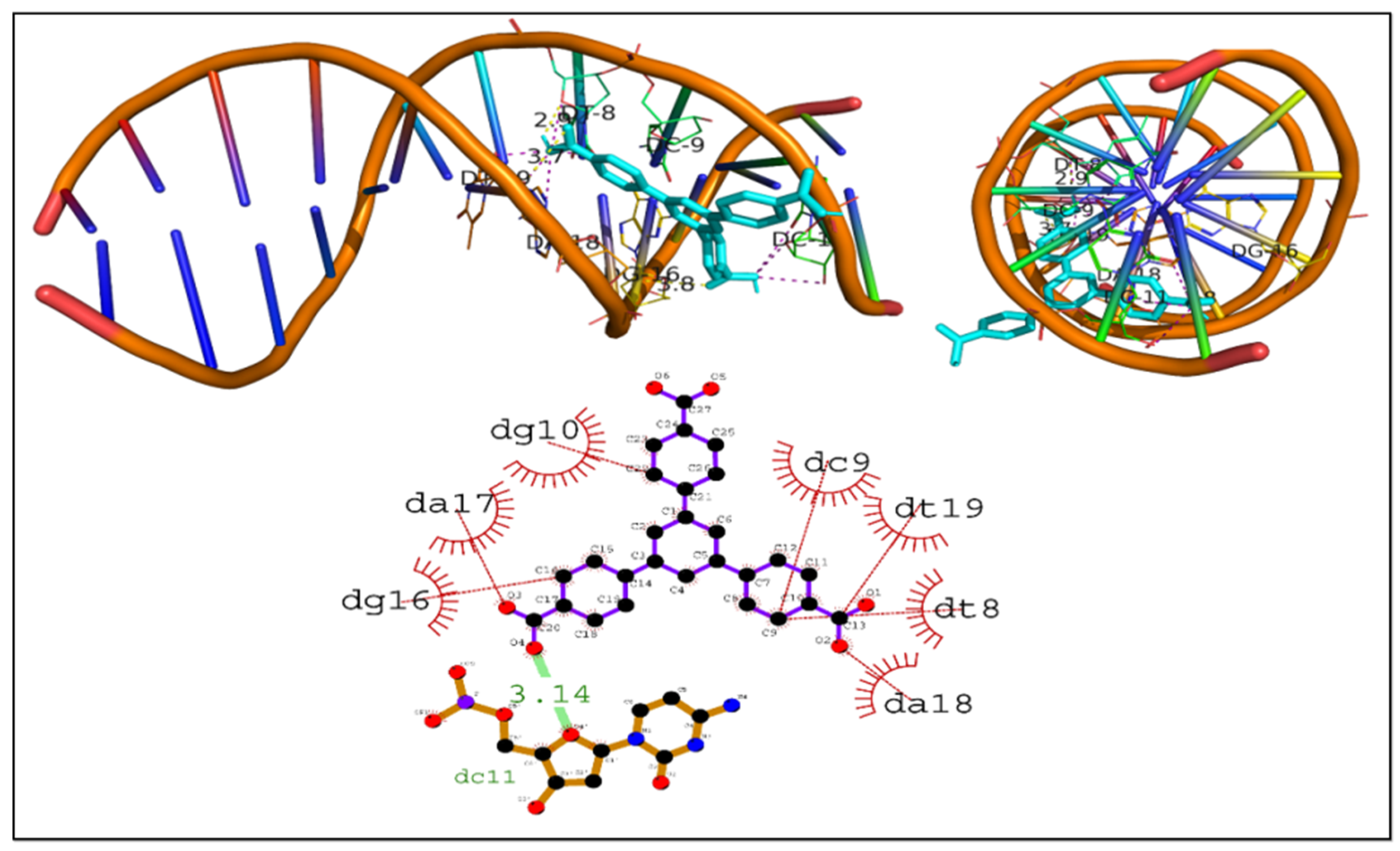
3.6.1. Intercalation of DNA Molecules
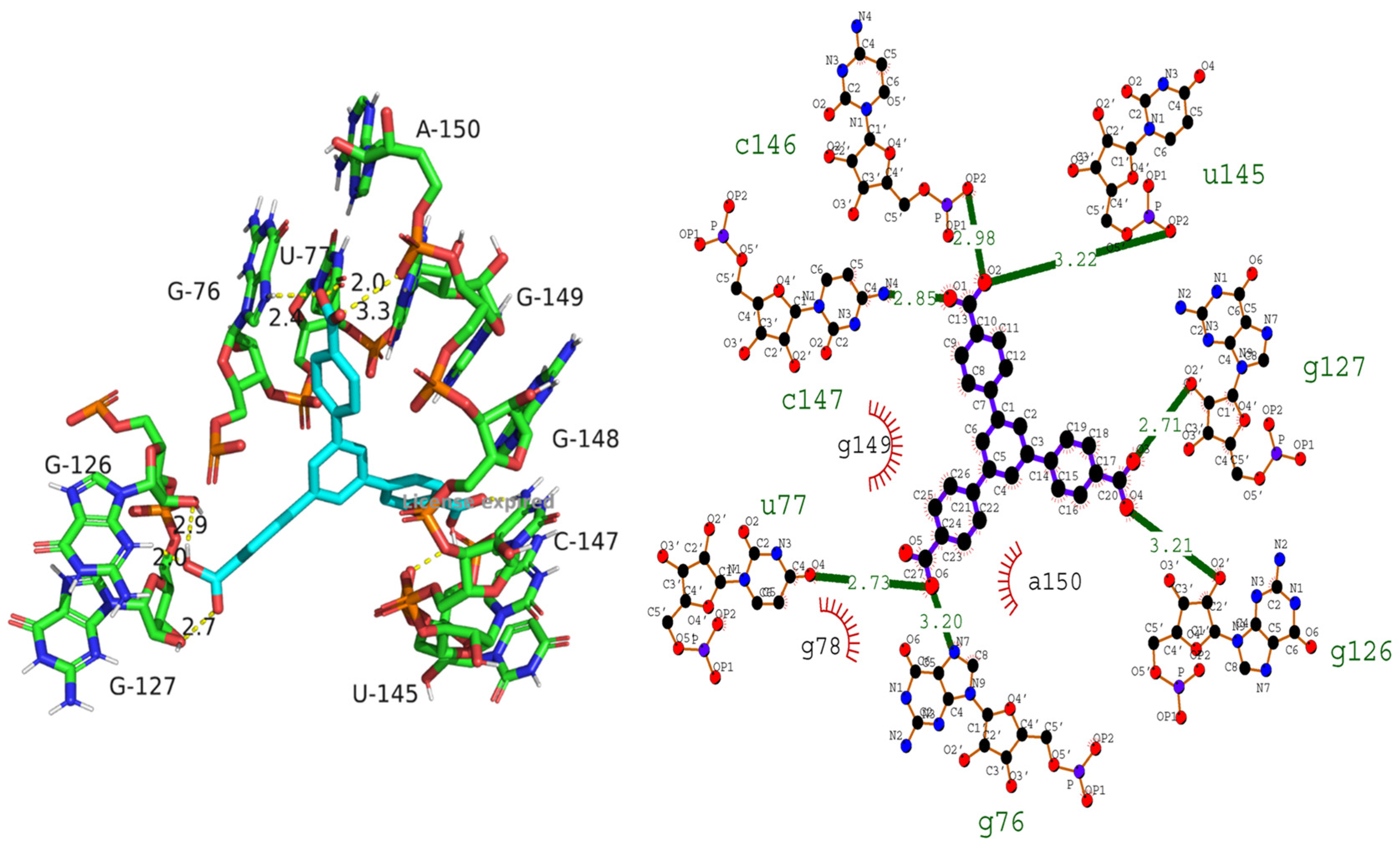
3.6.2. Intercalation of RNA Molecule
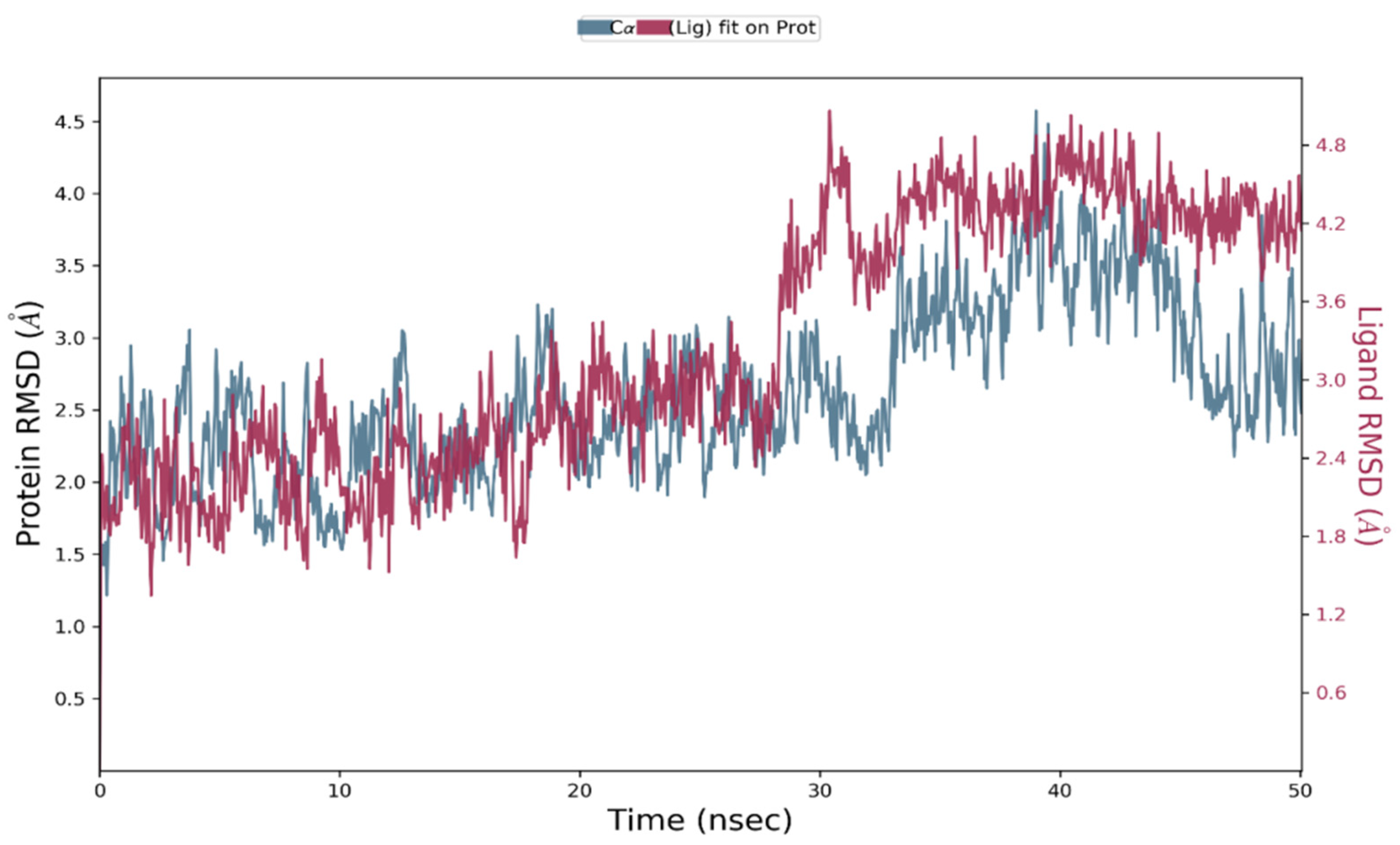
3.7. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Küpeli Akkol, E.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Feng, Y.; Panwar, N.; Tng, D.J.; Tjin, S.C.; Wang, K.; Yong, K.T. The application of mesoporous silica nanoparticle family in cancer theranostics. Coord. Chem. Chem. Rev. 2016, 319, 86–109. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141. [Google Scholar] [CrossRef]
- Gali-Muhtasib, H.; Hmadi, R.; Kareh, M.; Tohme, R.; Darwiche, N. Cell death mechanisms of plant-derived anticancer drugs: Beyond apoptosis. Apoptosis 2015, 20, 1531–1562. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.; Mir, M.I.; Perveen, F.; Javed, A.; Javaid, M.; Saeed, A.; Channar, P.A.; Farooqi, S.I.; Alkahtani, S.; Anwar, J. Investigations on anticancer potentials by DNA binding and cytotoxicity studies for newly synthesized and characterized imidazolidine and thiazolidine-based isatin derivatives. Molecules 2022, 27, 354. [Google Scholar] [CrossRef] [PubMed]
- Raghu, M.S.; Kumar, C.P.; Prashanth, M.K.; Kumar, K.Y.; Prathibha, B.S.; Kanthimathi, G.; Alissa, S.A.; Alghulikah, H.A.; Osman, S.M. Novel 1,3,5-triazine-based pyrazole derivatives as potential antitumor agents and EFGR kinase inhibitors: Synthesis, cytotoxicity, DNA binding, molecular docking and DFT studies. New J. Chem. 2021, 45, 13909–13924. [Google Scholar] [CrossRef]
- Arshad, N.; Abbas, N.; Perveen, F.; Mirza, B.; Almuhaini, A.M.; Alkahtani, S. Molecular docking analysis and spectroscopic investigations of zinc (II), nickel (II) N-phthaloyl-β-alanine complexes for DNA binding: Evaluation of antibacterial and antitumor activities. J. Saudi Chem. Soc. 2021, 25, 101323. [Google Scholar] [CrossRef]
- Nosrati, M.; Roushani, M. Three-dimensional modeling of streptomycin binding single-stranded DNA for aptamer-based biosensors, a molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2022, 1–10. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alsahli, M.A.; Almatroudi, A.; Verma, A.K.; Aloliqi, A.; Allemailem, K.S.; Khan, A.A.; Rahmani, A.H. Potential therapeutic targets of quercetin, a plant flavonol, and its role in the therapy of various types of cancer through the modulation of various cell signaling pathways. Molecules 2021, 26, 1315. [Google Scholar] [CrossRef]
- Haque, A.; Alam, Q.; Zubair Alam, M.; IAzhar, E.; Hussain Wali Sait, K.; Anfinan, N.; Mushtaq, G.; Amjad Kamal, M.; Rasool, M. Current understanding of HSP90 as a novel therapeutic target: An emerging approach for the treatment of cancer. Curr. Pharm. Des. 2016, 22, 2947–2959. [Google Scholar] [CrossRef]
- Heo, J.; Eki, R.; Abbas, T. Deregulation of F-box proteins and its consequence on cancer development, progression and metastasis. Semin. Cancer Biol. 2016, 36, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, H.; Wang, B.; Liao, Q.; Li, W.; Jin, X.; Cui, S.; Zhang, Y.; Ding, Y.; Zhao, L. miR-133a represses tumour growth and metastasis in colorectal cancer by targeting LIM and SH3 protein 1 and inhibiting the MAPK pathway. Eur. J. Cancer 2013, 49, 3924–3935. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- D’Amelio, M.; Sheng, M.; Cecconi, F. Caspase-3 in the central nervous system: Beyond apoptosis. Trends Neurosci. 2012, 35, 700–709. [Google Scholar] [CrossRef]
- Hong, B.; van den Heuvel, P.J.; VPrabhu, V.; Zhang, S.; SEl-Deiry, W. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef]
- Huang, J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol. Ther. 2021, 220, 107720. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, Y. Targeting p53 for novel anticancer therapy. Transl. Oncol. 2010, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Antonijevic, M.; Rochais, C.; Dallemagne, P. C3-Symmetric Ligands in Drug Design: When the Target Controls the Aesthetics of the Drug. Molecules 2023, 28, 679. [Google Scholar] [CrossRef] [PubMed]
- Agonigi, G.; Ciancaleoni, G.; Funaioli, T.; Zacchini, S.; Pineider, F.; Pinzino, C.; Pampaloni, G.; Zanotti, V.; Marchetti, F. Controlled Dissociation of Iron and Cyclopentadienyl from a Diiron Complex with a Bridging C3 Ligand Triggered by One-Electron Reduction. Inorg. Chem. 2018, 57, 15172–15186. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Hecker, M.; Koepp, E.; Orlia, W.; Czugler, M.; Csöregh, I. New trigonal lattice hosts: Stoicheiometric crystal inclusions of laterally trisubstituted benzenes—X-ray crystal structure of 1, 3, 5-tris-(4-carboxyphenyl) benzene dimethylformamide. J. Chem. Soc. Perkin Trans. 2 1988, 7, 1251–1257. [Google Scholar] [CrossRef]
- Kampschulte, L.; Lackinger, M.; Maier, A.K.; Kishore, R.S.; Griessl, S.; Schmittel, M.; Heckl, W.M. Solvent induced polymorphism in supramolecular 1, 3, 5-benzenetribenzoic acid monolayers. J. Phys. Chem. B 2006, 110, 10829–10836. [Google Scholar] [CrossRef] [PubMed]
- Khyazeva, M.K.; Fomkin, A.A.; Shkolin, A.V.; Men’shchikov, I.E.; Grinchenko, A.E.; Solovtsova, O.V.; Pulin, A.L. Adsorption Properties of a Functional Porous Material Based on a Zn–BTB Metal–Organic Framework Structure. Prot. Met. Phys. Chem. Surfaces. 2022, 58, 6–12. [Google Scholar] [CrossRef]
- Li, X.J.; Jiang, F.L.; Wu, M.Y.; Chen, L.; Qian, J.J.; Zhou, K.; Yuan, D.Q.; Hong, M.C. Construction of Two Microporous Metal–Organic Frameworks with flu and pyr Topologies Based on Zn4 (μ3-OH) 2 (CO2) 6 and Zn6 (μ6-O)(CO2) 6 Secondary Building Units. Inorg. Chemistry 2014, 53, 1032–1038. [Google Scholar] [CrossRef]
- Solovtsova, O.V.; Pulin, A.L.; Men’shchikov, I.E.; Platonova, N.P.; Knyazeva, M.K.; Chugaev, S.S.; Shkolin, A.V.; Fomkin, A.A. Zr-based metal–organic nanoporous adsorbents of high density for methane storage. Prot. Met. Phys. Chem. Surf. 2020, 56, 1114–1121. [Google Scholar] [CrossRef]
- Rad, A.S.; Ardjmand, M.; Esfahani, M.R.; Khodashenas, B. DFT calculations towards the geometry optimization, electronic structure, infrared spectroscopy and UV–vis analyses of Favipiravir adsorption on the first-row transition metals doped fullerenes; a new strategy for COVID-19 therapy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 247, 119082. [Google Scholar] [CrossRef]
- Bilal, M.S.; Ejaz, S.A.; Zargar, S.; Akhtar, N.; Wani, T.A.; Riaz, N.; Aborode, A.T.; Siddique, F.; Altwaijry, N.; Alkahtani, H.M.; et al. Computational Investigation of 1, 3, 4 Oxadiazole Derivatives as Lead Inhibitors of VEGFR 2 in Comparison with EGFR: Density Functional Theory, Molecular Docking and Molecular Dynamics Simulation Studies. Biomolecules 2022, 12, 1612. [Google Scholar] [CrossRef]
- Nikš, M.; Otto, M.; Bušová, B.; Štefanović, J. Quantification of proliferative and suppressive responses of human T lymphocytes following ConA stimulation. J. Immunol. Methods 1990, 126, 263–271. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Manojkumar, P.; Mahipal, V.; Suresh, G.; Venkatesh, N.; Ramesh, M.; Parthasarathy, T. Exploring Interaction Dynamics of Designed Organic Charge Transfer Complex of 6-Aminoindole and Chloranilic Acid: Spectrophotometric, Characterization, Computational, Antimicrobial, and DNA Binding Properties. J. Mol. Struct. 2022, 1258, 132666. [Google Scholar] [CrossRef]
- Patel, S.; Patel, P.; Undre, S.B.; Pandya, S.R.; Singh, M.; Bakshi, S. DNA binding and dispersion activities of titanium dioxide nanoparticles with UV/vis spectrophotometry, fluorescence spectroscopy and physicochemical analysis at physiological temperature. J. Mol. Liq. 2016, 213, 304–311. [Google Scholar] [CrossRef]
- Caricato, M.; Frisch, M.J.; Hiscocks, J.; Frisch, M.J. Gaussian 09: IOps Reference; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- St-Amant, A. Density functional methods in biomolecular modeling. Rev. Comput. Chem. 1996, 7, 217. [Google Scholar]
- Thanikaivelan, P.; Subramanian, V.; Rao, J.R.; Nair, B.U. Application of quantum chemical descriptor in quantitative structure activity and structure property relationship. Chem. Phys. Lett. 2000, 323, 59–70. [Google Scholar] [CrossRef]
- Baerends, E.J.; Gritsenko, O.V. A quantum chemical view of density functional theory. J. Phys. Chem. A 1997, 101, 5383–5403. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 365–382. [Google Scholar]
- Aziz, M.; Ejaz, S.A.; Tamam, N.; Siddique, F.; Riaz, N.; Qais, F.A.; Chtita, S.; Iqbal, J. Identification of potent inhibitors of NEK7 protein using a comprehensive computational approach. Sci. Rep. 2022, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 37. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.D.; Friesner., R.A.; Berne, B.J. A multiple-time-step molecular dynamics algorithm for macromolecules. J. Phys. Chem. A 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Luty, B.A.; Davis, M.E.; Tironi, I.G.; Van Gunsteren, W.F. A comparison of particle-particle, particle-mesh and Ewald methods for calculating electrostatic interactions in periodic molecular systems. Mol. Simul. 1994, 14, 11–20. [Google Scholar] [CrossRef]
- Fauzi, A.N.; Norazmi, M.N.; Yaacob, N.S. Tualang honey induces apoptosis and disrupts the mitochondrial membrane potential of human breast and cervical cancer cell lines. Food Chem. Toxicol. 2011, 49, 871–878. [Google Scholar] [CrossRef]
- Saeed, A.; Ejaz, S.A.; Saeed, M.; Channar, P.A.; Aziz, M.; Fayyaz, A.; Zargar, S.; Wani, T.A.; Alanazi, H.; Alharbi, M.; et al. Synthesis, Biochemical Characterization, and in-Silico Investigations of Acyl-3-(Ciprofloxacinyl) Thioureas as Inhibitors of Carbonic Anhydrase-II. Polycycl. Aromat. Compounds 2023, 1–19. [Google Scholar] [CrossRef]
- Kumar, S.; Nair, M.S. Deciphering the interaction of flavones with calf thymus DNA and octamer DNA sequence (CCAATTGG) 2. RSC Adv. 2021, 11, 29354–29371. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Z.; Zhou, M.; Li, Y. Synthesis and crystal structure of new monometallic Ni (II) and Co (II) complexes with an asymmetrical aroylhydrazone: Effects of the complexes on DNA/protein binding property, molecular docking, and in vitro anticancer activity. RSC Adv. 2017, 7, 49404–49422. [Google Scholar] [CrossRef]
- Zargar, S.; Wani, T.A.; Alsaif, N.A.; Khayyat, A.I.A. A Comprehensive Investigation of Interactions between Antipsychotic Drug Quetiapine and Human Serum Albumin Using Multi-Spectroscopic, Biochemical, and Molecular Modeling Approaches. Molecules 2022, 27, 2589. [Google Scholar] [CrossRef] [PubMed]
- Aleksić, M.M.; Kapetanović, V. An overview of the optical and electrochemical methods for detection of DNA–drug interactions. Acta Chim. Slov. 2014, 61, 555–573. [Google Scholar] [PubMed]
- Zargar, S.; Wani, T.A. Exploring the binding mechanism and adverse toxic effects of persistent organic pollutant (dicofol) to human serum albumin: A biophysical, biochemical and computational approach. Chem.-Biol. Interact. 2021, 350, 109707. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Al-Mehiziaa, A.A.; Bakheit, A.H.; Zargar, S.; Wani, T.A. A spectroscopic, thermodynamic and molecular docking study of the binding mechanism of dapoxetine with calf thymus DNA. S. Afr. J. Chem. 2020, 73, 44–50. [Google Scholar] [CrossRef]
- Li, X.L.; Hu, Y.J.; Wang, H.; Yu, B.Q.; Yue, H.L. Molecular spectroscopy evidence of berberine binding to DNA: Comparative binding and thermodynamic profile of intercalation. Biomacromolecules 2012, 13, 873–880. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound |  | |||
|---|---|---|---|---|
| HeLa | MDA-MB231 | MCF-7 | Vero Cells | |
| GI50 ± SEM | % Growth Inhibition | |||
| H3BTB | 16.2 ± 1.02 | 7.62 ± 0.91 | 9.03 ± 0.18 | 7.31% |
| Doxorubicin | 4.21 ± 0.22 | 6.82 ± 0.59 | 7.32 ± 0.81 | 10.2% |
| Cisplatin | 2.64 ± 0.13 | 2.27 ± 0.24 | 4.63 ± 0.21 | 6.67% |
| Compound | Optimization Energy (Hartree) | Polarizability a.u (α) | Dipole Moment (debye) | Potential Ionization I (eV) | Affinity A (eV) | Electron Donating Power (ω−) | Electron Accepting Power (ω+) | Electro Philicity (Δω±) |
|---|---|---|---|---|---|---|---|---|
| H3BTB | −1490.693 | 326.515 | 4.487 | 0.257 | 0.085 | 0.096 | 0.267 | 0.362 |
| Compound | EHOMO (eV) | ELUMO (eV) | ∆Egap (eV) | Chemical Hardness (η) | Chemical Potential (μ) | Electrophilicity Index (ω) | Chemical Softness (S) | Electronegativity (X) |
|---|---|---|---|---|---|---|---|---|
| H3BTB | −0.257 | −0.085 | 0.171 | 0.086 | −0.171 | 0.170 | 5.821 | 0.171 |
| Complex | Docking Score (kcal/mol) | Hydrogen Bonding Residues | Hydrogen Bond Length (Angstroms) | Hydrophobic Interactions Residues |
|---|---|---|---|---|
| Caspase-3–H3BTB | −10.4 | Gly60 | 2.88 | Cys170, Leu168, Thr255, His121, Tyr204, Gly122, Gly165, Thr166, Phe256, Leu168, Thr166, Phe256, and Thr62 |
| NF-κB–H3BTB | −8.8 | Phe239, Phe239, Gln266, and Thr257 | 3.02, 2.94, 3.14, and 3.21 | Gly259, Trp258, Gln241, Arg260, Ser238, Gly237, Glu222, and Lys28 |
| P53–H3BTB | −8.1 | Gln23, Gln23, Ser204, and Ala200 | 2.94, 3.03, 2.80, and 2.77 | Arg203, Leu125, Pro115, Tyr92, Ile22, Cys114, and Arg104 |
| DNA–H3BTB | −8.3 | Dc11 | 3.14 | Gd16, Da17, Dg10, Dc9, Dt19, Dt8, and Da18 |
| RNA–H3BTB | −9.5 | C146, C147, U77, U145, G127, G126, G76, and G78 | 2.98, 2.85, 2.73, 3.22, 2.71, 3.21, 3.20, and 2.73 | G149, G78, and A150 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wani, T.A.; Zargar, S. Molecular Spectroscopy Evidence of 1,3,5-Tris(4-carboxyphenyl)benzene Binding to DNA: Anticancer Potential along with the Comparative Binding Profile of Intercalation via Modeling Studies. Cells 2023, 12, 1120. https://doi.org/10.3390/cells12081120
Wani TA, Zargar S. Molecular Spectroscopy Evidence of 1,3,5-Tris(4-carboxyphenyl)benzene Binding to DNA: Anticancer Potential along with the Comparative Binding Profile of Intercalation via Modeling Studies. Cells. 2023; 12(8):1120. https://doi.org/10.3390/cells12081120
Chicago/Turabian StyleWani, Tanveer A., and Seema Zargar. 2023. "Molecular Spectroscopy Evidence of 1,3,5-Tris(4-carboxyphenyl)benzene Binding to DNA: Anticancer Potential along with the Comparative Binding Profile of Intercalation via Modeling Studies" Cells 12, no. 8: 1120. https://doi.org/10.3390/cells12081120
APA StyleWani, T. A., & Zargar, S. (2023). Molecular Spectroscopy Evidence of 1,3,5-Tris(4-carboxyphenyl)benzene Binding to DNA: Anticancer Potential along with the Comparative Binding Profile of Intercalation via Modeling Studies. Cells, 12(8), 1120. https://doi.org/10.3390/cells12081120