

Inflammation as a Regulator of the Airway Surface Liquid pH in Cystic Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
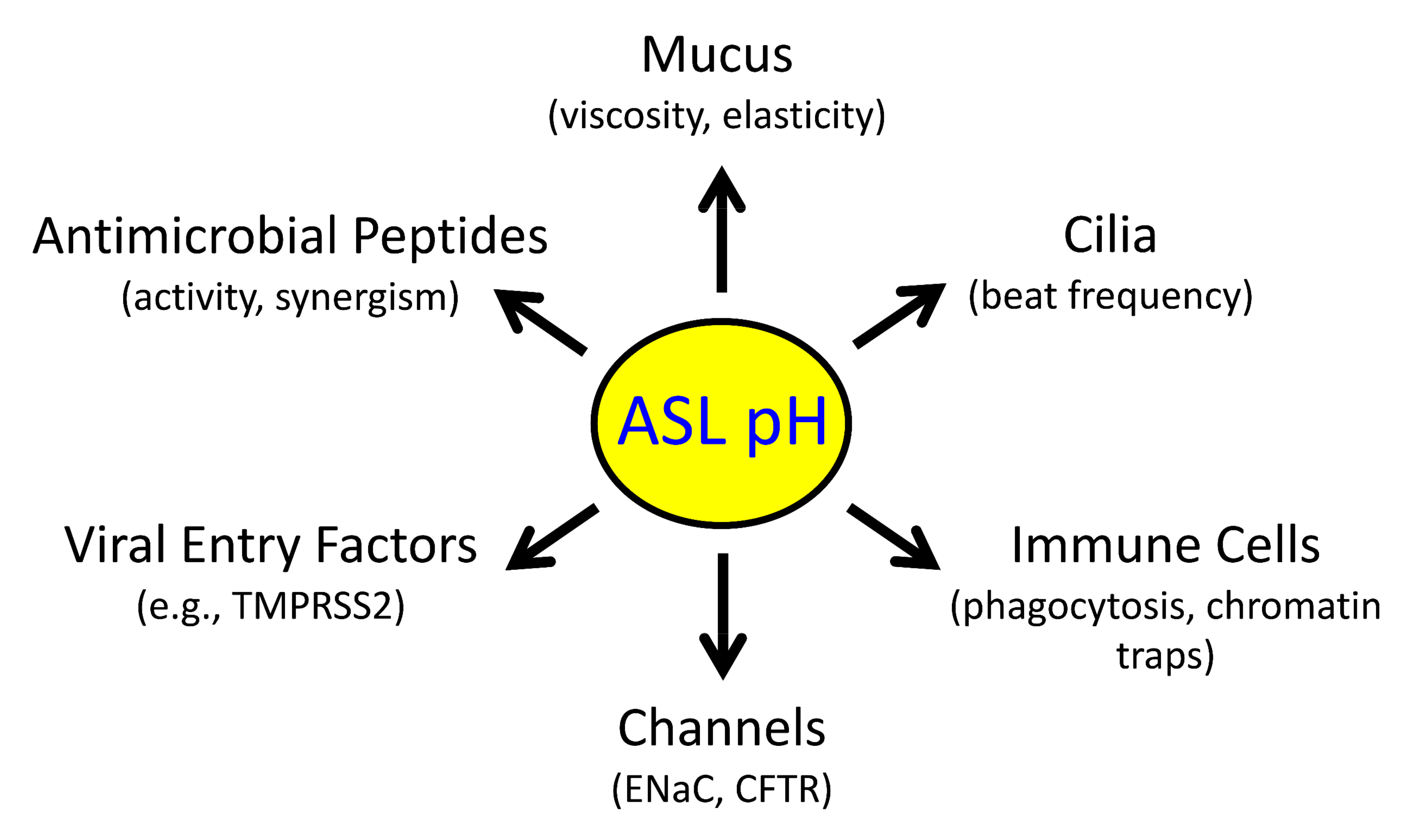
Abstract
1. Introduction
2. H+ and HCO3− Transporters Control pHASL
2.1. Reduced pHASL Disrupts Host Defenses
2.2. pHASL Changes with CF Airway Disease Progression
2.3. Inflammatory Cytokines Regulate pHASL
2.4. H+ Secretion
2.5. HCO3− Secretion
2.5.1. CFTR-Mediated HCO3− Secretion
2.5.2. Non-CFTR-Mediated HCO3− Secretion
2.5.3. Paracellular HCO3− Shunt
2.6. Other Regulatory Mechanisms
3. Airway Inflammation and CFTR Modulators Influence Each Other
4. Optimal Anti-Inflammatory Strategy in CF Is Unclear
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Widdicombe, J.H. Airway Epithelium; Morgan & Claypool: San Rafael, CA, USA, 2013. [Google Scholar]
- Widdicombe, J.H.; Widdicombe, J.G. Regulation of human airway surface liquid. Respir. Physiol. 1995, 99, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef]
- Laube, D.M.; Yim, S.; Ryan, L.K.; Kisich, K.O.; Diamond, G. Antimicrobial peptides in the airway. Curr. Top. Microbiol. Immunol. 2006, 306, 153–182. [Google Scholar] [CrossRef]
- Ermund, A.; Trillo-Muyo, S.; Hansson, G.C. Assembly, Release, and Transport of Airway Mucins in Pigs and Humans. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. S3), S159–S163. [Google Scholar] [CrossRef]
- Downey, D.G.; Bell, S.C.; Elborn, J.S. Neutrophils in cystic fibrosis. Thorax 2009, 64, 81–88. [Google Scholar] [CrossRef]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, M.A.; Fernandez, V.; Forteza, R.; Randell, S.H.; Salathe, M.; Conner, G.E. Transcellular thiocyanate transport by human airway epithelia. J. Physiol. 2004, 561 Pt 1, 183–194. [Google Scholar] [CrossRef]
- Moskwa, P.; Lorentzen, D.; Excoffon, K.J.; Zabner, J.; McCray, P.B., Jr.; Nauseef, W.M.; Dupuy, C.; Banfi, B. A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, E.R.; Tarran, R.; Grubb, B.R.; van Heusden, C.A.; Okada, S.; Boucher, R.C. Nucleotide release provides a mechanism for airway surface liquid homeostasis. J. Biol. Chem. 2004, 279, 36855–36864. [Google Scholar] [CrossRef] [PubMed]
- Van Heusden, C.; Grubb, B.R.; Button, B.; Lazarowski, E.R. Airway Epithelial Nucleotide Release Contributes to Mucociliary Clearance. Life 2021, 11, 430. [Google Scholar] [CrossRef]
- Berkebile, A.R.; Bartlett, J.A.; Abou Alaiwa, M.; Varga, S.M.; Power, U.F.; McCray, P.B., Jr. Airway Surface Liquid Has Innate Antiviral Activity That Is Reduced in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 104–111. [Google Scholar] [CrossRef]
- Laporte, M.; Naesens, L. Airway proteases: An emerging drug target for influenza and other respiratory virus infections. Curr. Opin. Virol. 2017, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Hill, D.B.; Button, B.; Rubinstein, M.; Boucher, R.C. Physiology and pathophysiology of human airway mucus. Physiol. Rev. 2022, 102, 1757–1836. [Google Scholar] [CrossRef]
- Voynow, J.A.; Rubin, B.K. Mucins, mucus, and sputum. Chest 2009, 135, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.K. Mucus structure and properties in cystic fibrosis. Paediatr. Respir. Rev. 2007, 8, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 1574–1575. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Quinton, P.M. Physiological basis of cystic fibrosis: A historical perspective. Physiol. Rev. 1999, 79 (Suppl. S1), S3–S22. [Google Scholar] [CrossRef]
- Smith, J.J.; Welsh, M.J. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J. Clin. Investig. 1992, 89, 1148–1153. [Google Scholar] [CrossRef]
- Poulsen, J.H.; Fischer, H.; Illek, B.; Machen, T.E. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 1994, 91, 5340–5344. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.X.; Ostedgaard, L.S.; Hoegger, M.J.; Moninger, T.O.; Karp, P.H.; McMenimen, J.D.; Choudhury, B.; Varki, A.; Stoltz, D.A.; Welsh, M.J. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J. Clin. Investig. 2016, 126, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjovall, H.; Hansson, G.C. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012, 209, 1263–1272. [Google Scholar] [CrossRef]
- Clary-Meinesz, C.; Mouroux, J.; Cosson, J.; Huitorel, P.; Blaive, B. Influence of external pH on ciliary beat frequency in human bronchi and bronchioles. Eur. Respir. J. 1998, 11, 330–333. [Google Scholar] [CrossRef]
- Khan, M.A.; Philip, L.M.; Cheung, G.; Vadakepeedika, S.; Grasemann, H.; Sweezey, N.; Palaniyar, N. Regulating NETosis: Increasing pH Promotes NADPH Oxidase-Dependent NETosis. Front. Med. 2018, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Garland, A.L.; Walton, W.G.; Coakley, R.D.; Tan, C.D.; Gilmore, R.C.; Hobbs, C.A.; Tripathy, A.; Clunes, L.A.; Bencharit, S.; Stutts, M.J.; et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2013, 110, 15973–15978. [Google Scholar] [CrossRef]
- Abou Alaiwa, M.H.; Reznikov, L.R.; Gansemer, N.D.; Sheets, K.A.; Horswill, A.R.; Stoltz, D.A.; Zabner, J.; Welsh, M.J. pH modulates the activity and synergism of the airway surface liquid antimicrobials beta-defensin-3 and LL-37. Proc. Natl. Acad. Sci. USA 2014, 111, 18703–18708. [Google Scholar] [CrossRef]
- Middleton, P.G.; Taylor-Cousar, J.L. Development of elexacaftor–tezacaftor–ivacaftor: Highly effective CFTR modulation for the majority of people with Cystic Fibrosis. Expert Rev. Respir. Med. 2021, 15, 723–735. [Google Scholar] [CrossRef]
- Myerburg, M.; Pilewski, J.M. CFTR Modulators to the Rescue of Individuals with Cystic Fibrosis and Advanced Lung Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 7–9. [Google Scholar] [CrossRef]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lam, C.S.; Wu, F.; Wang, W.; Duan, Y.; Huang, P. Regulation of CFTR channels by HCO(3)--sensitive soluble adenylyl cyclase in human airway epithelial cells. Am. J. Physiol. Cell Physiol. 2005, 289, C1145–C1151. [Google Scholar] [CrossRef]
- Baudouin-Legros, M.; Hamdaoui, N.; Borot, F.; Fritsch, J.; Ollero, M.; Planelles, G.; Edelman, A. Control of basal CFTR gene expression by bicarbonate-sensitive adenylyl cyclase in human pulmonary cells. Cell. Physiol. Biochem. 2008, 21, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberger, A.J.B.; Sanyal, A.; Saminathan, A.; Bloyet, L.M.; Stumpf, S.; Liu, Z.; Ojha, R.; Patjas, M.T.; Geneid, A.; Scanavachi, G.; et al. SARS-CoV-2 requires acidic pH to infect cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2209514119. [Google Scholar] [CrossRef]
- Zajac, M.; Dreano, E.; Edwards, A.; Planelles, G.; Sermet-Gaudelus, I. Airway Surface Liquid pH Regulation in Airway Epithelium Current Understandings and Gaps in Knowledge. Int. J. Mol. Sci. 2021, 22, 3384. [Google Scholar] [CrossRef]
- Fischer, H.; Widdicombe, J.H. Mechanisms of acid and base secretion by the airway epithelium. J. Membr. Biol. 2006, 211, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Rehman, T.; Karp, P.H.; Tan, P.; Goodell, B.J.; Pezzulo, A.A.; Thurman, A.L.; Thornell, I.M.; Durfey, S.L.; Duffey, M.E.; Stoltz, D.A.; et al. Inflammatory cytokines TNF-alpha and IL-17 enhance the efficacy of cystic fibrosis transmembrane conductance regulator modulators. J. Clin. Investig. 2021, 131, e150398. [Google Scholar] [CrossRef] [PubMed]
- Rehman, T.; Thornell, I.M.; Pezzulo, A.A.; Thurman, A.L.; Romano Ibarra, G.S.; Karp, P.H.; Tan, P.; Duffey, M.E.; Welsh, M.J. TNFalpha and IL-17 alkalinize airway surface liquid through CFTR and pendrin. Am. J. Physiol. Cell Physiol. 2020, 319, C331–C344. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Villacreses, R.; Thornell, I.M.; Noriega, J.; Mather, S.; Brommel, C.M.; Lu, L.; Zabner, A.; Ehler, A.; Meyerholz, D.K.; et al. V-Type ATPase Mediates Airway Surface Liquid Acidification in Pig Small Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2021, 65, 146–156. [Google Scholar] [CrossRef]
- Shamsuddin, A.K.; Quinton, P.M. Native small airways secrete bicarbonate. Am. J. Respir. Cell Mol. Biol. 2014, 50, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J. Mechanisms of bicarbonate secretion: Lessons from the airways. Cold Spring Harb. Perspect. Med. 2012, 2, a015016. [Google Scholar] [CrossRef] [PubMed]
- Devor, D.C.; Singh, A.K.; Lambert, L.C.; DeLuca, A.; Frizzell, R.A.; Bridges, R.J. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J. Gen. Physiol. 1999, 113, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Kreindler, J.L.; Peters, K.W.; Frizzell, R.A.; Bridges, R.J. Identification and membrane localization of electrogenic sodium bicarbonate cotransporters in Calu-3 cells. Biochim. Biophys. Acta 2006, 1762, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases—An overview. Curr. Pharm. Des. 2008, 14, 603–614. [Google Scholar] [CrossRef]
- Gorrieri, G.; Scudieri, P.; Caci, E.; Schiavon, M.; Tomati, V.; Sirci, F.; Napolitano, F.; Carrella, D.; Gianotti, A.; Musante, I.; et al. Goblet Cell Hyperplasia Requires High Bicarbonate Transport To Support Mucin Release. Sci. Rep. 2016, 6, 36016. [Google Scholar] [CrossRef]
- Alper, S.L. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J. Exp. Biol. 2009, 212 Pt 11, 1672–1683. [Google Scholar] [CrossRef]
- Hollenhorst, M.I.; Richter, K.; Fronius, M. Ion transport by pulmonary epithelia. J. Biomed. Biotechnol. 2011, 2011, 174306. [Google Scholar] [CrossRef]
- Xu, H.; Ghishan, F.K.; Kiela, P.R. SLC9 Gene Family: Function, Expression, and Regulation. Compr. Physiol. 2018, 8, 555–583. [Google Scholar] [CrossRef]
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Pranke, I.; Villeret, B.; Cottart, C.H.; et al. Airway surface liquid acidification initiates host defense abnormalities in Cystic Fibrosis. Sci. Rep. 2019, 9, 6516. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Guequen, A.; Philp, A.R.; Villanueva, S.; Apablaza, T.; Fernandez-Moncada, I.; Mansilla, A.; Delpiano, L.; Ruminot, I.; Carrasco, C.; et al. Inhibition of the sodium-dependent HCO3(-) transporter SLC4A4, produces a cystic fibrosis-like airway disease phenotype. eLife 2022, 11, e75871. [Google Scholar] [CrossRef]
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34. [Google Scholar] [CrossRef]
- McCarron, A.; Donnelley, M.; Parsons, D. Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res. 2018, 19, 54. [Google Scholar] [CrossRef]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Vecchio-Pagan, B.; Sharma, N.; Waheed, A.; Li, X.; Raraigh, K.S.; Robbins, S.; Han, S.T.; Franca, A.L.; Pellicore, M.J.; et al. Loss of carbonic anhydrase XII function in individuals with elevated sweat chloride concentration and pulmonary airway disease. Hum. Mol. Genet. 2016, 25, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Liao, J.; Scales, N.B.; Martini, C.; Luan, X.; Abu-Arish, A.; Robert, R.; Luo, Y.; McKay, G.A.; Nguyen, D.; et al. Large pH oscillations promote host defense against human airways infection. J. Exp. Med. 2021, 218, e20201831. [Google Scholar] [CrossRef]
- Kim, D.; Liao, J.; Hanrahan, J.W. The buffer capacity of airway epithelial secretions. Front. Physiol. 2014, 5, 188. [Google Scholar] [CrossRef]
- Holma, B. Influence of buffer capacity and pH-dependent rheological properties of respiratory mucus on health effects due to acidic pollution. Sci. Total Environ. 1985, 41, 101–123. [Google Scholar] [CrossRef]
- Holma, B.; Hegg, P.O. pH- and protein-dependent buffer capacity and viscosity of respiratory mucus. Their interrelationships and influence on health. Sci. Total Environ. 1989, 84, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O'Neal, W.K.; Boucher, R.C. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef]
- Tate, S.; MacGregor, G.; Davis, M.; Innes, J.A.; Greening, A.P. Airways in cystic fibrosis are acidified: Detection by exhaled breath condensate. Thorax 2002, 57, 926–929. [Google Scholar] [CrossRef]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A.S. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef]
- McShane, D.; Davies, J.C.; Davies, M.G.; Bush, A.; Geddes, D.M.; Alton, E.W. Airway surface pH in subjects with cystic fibrosis. Eur. Respir. J. 2003, 21, 37–42. [Google Scholar] [CrossRef]
- Schultz, A.; Puvvadi, R.; Borisov, S.M.; Shaw, N.C.; Klimant, I.; Berry, L.J.; Montgomery, S.T.; Nguyen, T.; Kreda, S.M.; Kicic, A.; et al. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat. Commun. 2017, 8, 1409. [Google Scholar] [CrossRef]
- Sly, P.D.; Brennan, S.; Gangell, C.; de Klerk, N.; Murray, C.; Mott, L.; Stick, S.M.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 2009, 180, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar] [CrossRef]
- Balough, K.; McCubbin, M.; Weinberger, M.; Smits, W.; Ahrens, R.; Fick, R. The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr. Pulmonol. 1995, 20, 63–70. [Google Scholar] [CrossRef]
- Ranganathan, S.C.; Hall, G.L.; Sly, P.D.; Stick, S.M.; Douglas, T.A.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF). Early Lung Disease in Infants and Preschool Children with Cystic Fibrosis. What Have We Learned and What Should We Do about It? Am. J. Respir. Crit. Care Med. 2017, 195, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Abou Alaiwa, M.H.; Beer, A.M.; Pezzulo, A.A.; Launspach, J.L.; Horan, R.A.; Stoltz, D.A.; Starner, T.D.; Welsh, M.J.; Zabner, J. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J. Cyst. Fibros. 2014, 13, 373–377. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2010, 2, 29ra31. [Google Scholar] [CrossRef]
- Deschamp, A.R.; Hatch, J.E.; Slaven, J.E.; Gebregziabher, N.; Storch, G.; Hall, G.L.; Stick, S.; Ranganathan, S.; Ferkol, T.W.; Davis, S.D. Early respiratory viral infections in infants with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 844–850. [Google Scholar] [CrossRef]
- Xu, W.; Zheng, S.; Goggans, T.M.; Kiser, P.; Quinones-Mateu, M.E.; Janocha, A.J.; Comhair, S.A.; Slee, R.; Williams, B.R.; Erzurum, S.C. Cystic fibrosis and normal human airway epithelial cell response to influenza a viral infection. J. Interferon Cytokine Res. 2006, 26, 609–627. [Google Scholar] [CrossRef]
- Zheng, S.; De, B.P.; Choudhary, S.; Comhair, S.A.; Goggans, T.; Slee, R.; Williams, B.R.; Pilewski, J.; Haque, S.J.; Erzurum, S.C. Impaired innate host defense causes susceptibility to respiratory virus infections in cystic fibrosis. Immunity 2003, 18, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Xu, W.; Bose, S.; Banerjee, A.K.; Haque, S.J.; Erzurum, S.C. Impaired nitric oxide synthase-2 signaling pathway in cystic fibrosis airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L374–L381. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.I.; Schaller-Bals, S.; Paul, K.P.; Wahn, U.; Bals, R. Beta-defensins and LL-37 in bronchoalveolar lavage fluid of patients with cystic fibrosis. J. Cyst. Fibros. 2004, 3, 45–50. [Google Scholar] [CrossRef]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schroder, J.M.; Wang, J.M.; Howard, O.M.; et al. Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef]
- Scott, A.; Weldon, S.; Buchanan, P.J.; Schock, B.; Ernst, R.K.; McAuley, D.F.; Tunney, M.M.; Irwin, C.R.; Elborn, J.S.; Taggart, C.C. Evaluation of the ability of LL-37 to neutralise LPS in vitro and ex vivo. PLoS ONE 2011, 6, e26525. [Google Scholar] [CrossRef]
- McAleer, J.P.; Kolls, J.K. Mechanisms controlling Th17 cytokine expression and host defense. J. Leukoc. Biol. 2011, 90, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Stoppelenburg, A.J.; Salimi, V.; Hennus, M.; Plantinga, M.; Huis in 't Veld, R.; Walk, J.; Meerding, J.; Coenjaerts, F.; Bont, L.; Boes, M. Local IL-17A potentiates early neutrophil recruitment to the respiratory tract during severe RSV infection. PLoS ONE 2013, 8, e78461. [Google Scholar] [CrossRef]
- McAllister, F.; Henry, A.; Kreindler, J.L.; Dubin, P.J.; Ulrich, L.; Steele, C.; Finder, J.D.; Pilewski, J.M.; Carreno, B.M.; Goldman, S.J.; et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: Implications for airway inflammation in cystic fibrosis. J. Immunol. 2005, 175, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Karpati, F.; Hjelte, F.L.; Wretlind, B. TNF-alpha and IL-8 in consecutive sputum samples from cystic fibrosis patients during antibiotic treatment. Scand. J. Infect. Dis. 2000, 32, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Huang, J.; Billet, A.; Abu-Arish, A.; Goepp, J.; Matthes, E.; Tewfik, M.A.; Frenkiel, S.; Hanrahan, J.W. Pendrin Mediates Bicarbonate Secretion and Enhances Cystic Fibrosis Transmembrane Conductance Regulator Function in Airway Surface Epithelia. Am. J. Respir. Cell Mol. Biol. 2019, 60, 705–716. [Google Scholar] [CrossRef]
- Lennox, A.T.; Coburn, S.L.; Leech, J.A.; Heidrich, E.M.; Kleyman, T.R.; Wenzel, S.E.; Pilewski, J.M.; Corcoran, T.E.; Myerburg, M.M. ATP12A promotes mucus dysfunction during Type 2 airway inflammation. Sci. Rep. 2018, 8, 2109. [Google Scholar] [CrossRef]
- Haggie, P.M.; Phuan, P.W.; Tan, J.A.; Zlock, L.; Finkbeiner, W.E.; Verkman, A.S. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J. 2016, 30, 2187–2197. [Google Scholar] [CrossRef]
- Scudieri, P.; Musante, I.; Caci, E.; Venturini, A.; Morelli, P.; Walter, C.; Tosi, D.; Palleschi, A.; Martin-Vasallo, P.; Sermet-Gaudelus, I.; et al. Increased expression of ATP12A proton pump in cystic fibrosis airways. JCI Insight 2018, 3, e123616. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Young, L.; Chen, Y.; Ran, H.; Meyers, M.; Joseph, P.; Cho, Y.H.; Hassett, D.J.; Lau, G.W. Pseudomonas aeruginosa pyocyanin inactivates lung epithelial vacuolar ATPase-dependent cystic fibrosis transmembrane conductance regulator expression and localization. Cell. Microbiol. 2006, 8, 1121–1133. [Google Scholar] [CrossRef]
- Ran, H.; Hassett, D.J.; Lau, G.W. Human targets of Pseudomonas aeruginosa pyocyanin. Proc. Natl. Acad. Sci. USA 2003, 100, 14315–14320. [Google Scholar] [CrossRef]
- Garnett, J.P.; Kalsi, K.K.; Sobotta, M.; Bearham, J.; Carr, G.; Powell, J.; Brodlie, M.; Ward, C.; Tarran, R.; Baines, D.L. Hyperglycaemia and Pseudomonas aeruginosa acidify cystic fibrosis airway surface liquid by elevating epithelial monocarboxylate transporter 2 dependent lactate-H(+) secretion. Sci. Rep. 2016, 6, 37955. [Google Scholar] [CrossRef]
- Simoes, F.B.; Kmit, A.; Amaral, M.D. Cross-talk of inflammatory mediators and airway epithelium reveals the cystic fibrosis transmembrane conductance regulator as a major target. ERJ Open Res. 2021, 7, 00247–2021. [Google Scholar] [CrossRef]
- Gray, T.; Coakley, R.; Hirsh, A.; Thornton, D.; Kirkham, S.; Koo, J.S.; Burch, L.; Boucher, R.; Nettesheim, P. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1beta in human bronchial epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L320–L330. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, S.M.; Cihil, K.M.; Cornuet, P.K.; Myerburg, M.M.; Swiatecka-Urban, A. Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS ONE 2013, 8, e63167. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.D.; Bengtson, C.D.; Yoshida, M.; Niloy, A.J.; Dennis, J.S.; Baumlin, N.; Salathe, M. Losartan ameliorates TGF-beta1-induced CFTR dysfunction and improves correction by cystic fibrosis modulator therapies. J. Clin. Investig. 2022, 132, e155241. [Google Scholar] [CrossRef]
- Adams, K.M.; Abraham, V.; Spielman, D.; Kolls, J.K.; Rubenstein, R.C.; Conner, G.E.; Cohen, N.A.; Kreindler, J.L. IL-17A induces Pendrin expression and chloride-bicarbonate exchange in human bronchial epithelial cells. PLoS ONE 2014, 9, e103263. [Google Scholar] [CrossRef]
- Gray, M.A. Bicarbonate secretion: It takes two to tango. Nat. Cell Biol. 2004, 6, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Thornell, I.M.; Rehman, T.; Pezzulo, A.A.; Welsh, M.J. Paracellular bicarbonate flux across human cystic fibrosis airway epithelia tempers changes in airway surface liquid pH. J. Physiol. 2020, 598, 4307–4320. [Google Scholar] [CrossRef]
- Parker, M.D. Soda stream modifies airway fluid. J. Physiol. 2020, 598, 4143–4144. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Ohana, E.; Park, H.W.; Yang, D.; Muallem, S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 2012, 92, 39–74. [Google Scholar] [CrossRef]
- Rehman, T.; Karp, P.H.; Thurman, A.L.; Mather, S.E.; Jain, A.; Cooney, A.L.; Sinn, P.L.; Pezzulo, A.A.; Duffey, M.E.; Welsh, M.J. WNK Inhibition Increases Surface Liquid pH and Host Defense in Cystic Fibrosis Airway Epithelia. Am. J. Respir. Cell Mol. Biol. 2022, 67, 491–502. [Google Scholar] [CrossRef]
- Luscher, B.P.; Vachel, L.; Ohana, E.; Muallem, S. Cl(-) as a bona fide signaling ion. Am. J. Physiol. Cell Physiol. 2020, 318, C125–C136. [Google Scholar] [CrossRef]
- Shcheynikov, N.; Son, A.; Hong, J.H.; Yamazaki, O.; Ohana, E.; Kurtz, I.; Shin, D.M.; Muallem, S. Intracellular Cl- as a signaling ion that potently regulates Na+/HCO3- transporters. Proc. Natl. Acad. Sci. USA 2015, 112, E329–E337. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Mall, M.A.; Alvarez, A.; Colombo, C.; de Winter-de Groot, K.M.; Fajac, I.; McBennett, K.A.; McKone, E.F.; Ramsey, B.W.; Sutharsan, S.; et al. Triple Therapy for Cystic Fibrosis Phe508del-Gating and -Residual Function Genotypes. N. Engl. J. Med. 2021, 385, 815–825. [Google Scholar] [CrossRef]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 64–73. [Google Scholar] [CrossRef]
- Martin, C.; Legeai, C.; Regard, L.; Cantrelle, C.; Dorent, R.; Carlier, N.; Kerbaul, F.; Burgel, P.R. Major Decrease in Lung Transplantation for Patients with Cystic Fibrosis in France. Am. J. Respir. Crit. Care Med. 2022, 205, 584–586. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef] [PubMed]
- McNally, P.; Butler, D.; Karpievitch, Y.V.; Linnane, B.; Ranganathan, S.; Stick, S.M.; Hall, G.L.; Schultz, A. Ivacaftor and Airway Inflammation in Preschool Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Martino, M.E.B.; Minges, J.T.; Boyles, S.E.; Guhr Lee, T.N.; Esther, C.R., Jr.; Ribeiro, C.M.P. Airway Epithelial Inflammation In Vitro Augments the Rescue of Mutant CFTR by Current CFTR Modulator Therapies. Front. Pharmacol. 2021, 12, 628722. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Gentzsch, M. Impact of Airway Inflammation on the Efficacy of CFTR Modulators. Cells 2021, 10, 3260. [Google Scholar] [CrossRef]
- Keown, K.; Brown, R.; Doherty, D.F.; Houston, C.; McKelvey, M.C.; Creane, S.; Linden, D.; McAuley, D.F.; Kidney, J.C.; Weldon, S.; et al. Airway Inflammation and Host Responses in the Era of CFTR Modulators. Int. J. Mol. Sci. 2020, 21, 6379. [Google Scholar] [CrossRef]
- Harvey, C.; Weldon, S.; Elborn, S.; Downey, D.G.; Taggart, C. The Effect of CFTR Modulators on Airway Infection in Cystic Fibrosis. Int. J. Mol. Sci. 2022, 23, 3513. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef]
- Bitam, S.; Pranke, I.; Hollenhorst, M.; Servel, N.; Moquereau, C.; Tondelier, D.; Hatton, A.; Urbach, V.; Sermet-Gaudelus, I.; Hinzpeter, A.; et al. An unexpected effect of TNF-alpha on F508del-CFTR maturation and function. F1000Research 2015, 4, 218. [Google Scholar] [CrossRef]
- Lands, L.C.; Dauletbaev, N. High-Dose Ibuprofen in Cystic Fibrosis. Pharmaceuticals 2010, 3, 2213–2224. [Google Scholar] [CrossRef]
- Lands, L.C.; Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019, 9, CD001505. [Google Scholar] [CrossRef]
- Konstan, M.W.; Byard, P.J.; Hoppel, C.L.; Davis, P.B. Effect of high-dose ibuprofen in patients with cystic fibrosis. N. Engl. J. Med. 1995, 332, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Mueller, G.; Hadjiliadis, D.; Hoag, J.B.; Lubsch, L.; Hazle, L.; Sabadosa, K.; Marshall, B.; et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2013, 187, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Urbach, V.; Verriere, V.; Grumbach, Y.; Bousquet, J.; Harvey, B.J. Rapid anti-secretory effects of glucocorticoids in human airway epithelium. Steroids 2006, 71, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Bossmann, M.; Thome, U.H. Glucocorticoids Distinctively Modulate the CFTR Channel with Possible Implications in Lung Development and Transition into Extrauterine Life. PLoS ONE 2015, 10, e0124833. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, T.; Welsh, M.J. Inflammation as a Regulator of the Airway Surface Liquid pH in Cystic Fibrosis. Cells 2023, 12, 1104. https://doi.org/10.3390/cells12081104
Rehman T, Welsh MJ. Inflammation as a Regulator of the Airway Surface Liquid pH in Cystic Fibrosis. Cells. 2023; 12(8):1104. https://doi.org/10.3390/cells12081104
Chicago/Turabian StyleRehman, Tayyab, and Michael J. Welsh. 2023. "Inflammation as a Regulator of the Airway Surface Liquid pH in Cystic Fibrosis" Cells 12, no. 8: 1104. https://doi.org/10.3390/cells12081104
APA StyleRehman, T., & Welsh, M. J. (2023). Inflammation as a Regulator of the Airway Surface Liquid pH in Cystic Fibrosis. Cells, 12(8), 1104. https://doi.org/10.3390/cells12081104