


Current Strategy to Treat Immunogenic Gastrointestinal Cancers: Perspectives for a New Era



Abstract
1. Results of Early-Phase Clinical Trials Were Disappointing for Colorectal Cancers
2. Opening the Door to the Immune-Oncology Field

{kind=link}
{kind=link}
{kind=link}
| Study | KEYNOTE-164 [21] | KEYNOTE-177 [23] | CheckMate-142 [24,25,26] | ||||
|---|---|---|---|---|---|---|---|
| Treatment line | ≥2nd line | ≥3rd line | 1st line | ≥2nd line | ≥2nd line | 1st line | |
| Phase | II | III | II | ||||
| No. of patients | 63 | 61 | 153 | 154 | 74 | 119 | 45 |
| Primary endpoint | ORR | PFS and OS | ORR | ||||
| Regimen | Pembrolizumab 200 mg every 3 weeks | Pembrolizumab 200 mg every 3 weeks | Chemotherapy | Nivolumab 3 mg/kg every 2 weeks | Nivolumab 3 mg/kg plus ipilimumab 1 mg/kg every 3 weeks (4 cycles) followed by nivolumab 3 mg/kg every 2 weeks | Nivolumab 3 mg/kg every 2 weeks plus ipilimumab 1 mg/kg every 6 weeks | |
| ORR | 33% | 33% | 43.8% | 33.1% | 32% | 49% | 69% |
| DCR | 57% | 51% | 64.7% | 75.3% | 64% | 79% | 84% |
| Median PFS | 4.1 months | 2.3 months | 16.5 months | 8.2 months | 14.3 months | NR | NR |
| 12-month PFS rate | 41% | 34% | 55.3% | 37.3% | 50% | 71% | 76.4% |
| Median OS | NR | 31.4 months | NR | 36.7 months | NR | NR | NR |
| 12-month OS rate | 77% | 72% | 78% | 74% | 73% | 85% | 84.1% |
3. Current Status of Gastrointestinal (GI) Cancers with MSI-H/dMMR in Clinical Practice
3.1. CRC
3.2. Gastric/Gastroesophageal Junction Cancers (GC/GEJC)
3.3. Other GI Cancers
4. Next Challenge for Further Improvement in Patients with MSI-H/dMMR
5. An Avenue for Innovative Immunotherapy for MSI-H/dMMR GI Cancers: From Care to Cure
5.1. CRC
5.2. GC/GEJC
6. Future Perspectives on MSI-H/dMMR Cancers
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Schroering, A.G.; Edelbrock, M.A.; Richards, T.J.; Williams, K.J. The cell cycle and DNA mismatch repair. Exp. Cell Res. 2007, 313, 292–304. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef]
- Eso, Y.; Shimizu, T.; Takeda, H.; Takai, A.; Marusawa, H. Microsatellite instability and immune checkpoint inhibitors: Toward precision medicine against gastrointestinal and hepatobiliary cancers. J. Gastroenterol. 2020, 55, 15–26. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818. [Google Scholar] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A., Jr. Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. Oncologist 2016, 21, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.C.; Lin, M.T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Saeterdal, I.; Bjorheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Moller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jager, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Lenz, H.J.; Van Cutsem, E.; Luisa Limon, M.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2021, 40, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Hewish, M.; Lord, C.J.; Martin, S.A.; Cunningham, D.; Ashworth, A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat. Rev. Clin. Oncol. 2010, 7, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, K.; Shitoh, K.; Miyakura, Y.; Furukawa, T.; Yamashita, Y.; Ota, J.; Ohki, R.; Choi, Y.L.; Wada, T.; Konishi, F.; et al. Mutations of BRAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int. J. Cancer 2004, 108, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Sueur, B.; Zaanan, A.; de la Fouchardiere, C.; Sefrioui, D.; Lecomte, T.; Aparicio, T.; Des Guetz, G.; Artru, P.; Hautefeuille, V.; et al. Prognosis and chemosensitivity of deficient MMR phenotype in patients with metastatic colorectal cancer: An AGEO retrospective multicenter study. Int. J. Cancer 2020, 147, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Casak, S.J.; Marcus, L.; Fashoyin-Aje, L.; Mushti, S.L.; Cheng, J.; Shen, Y.L.; Pierce, W.F.; Her, L.; Goldberg, K.B.; Theoret, M.R.; et al. FDA Approval Summary: Pembrolizumab for the First-line Treatment of Patients with MSI-H/dMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021, 27, 4680–4684. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): Final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Bendell, J.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Peltola, K.; Jaeger, D.; Evans, J.; et al. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients With Metastatic Esophagogastric Cancer. J. Clin. Oncol. 2018, 36, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Nam, K.H.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Kim, S.H.; Chang, H.; Lee, J.O.; Kim, Y.J.; Lee, H.S.; et al. Prognostic implications of immunosuppressive protein expression in tumors as well as immune cell infiltration within the tumor microenvironment in gastric cancer. Gastric Cancer 2016, 19, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.; Li, Q.; Ren, T.; Xia, W.; Peng, Y.; Liu, G.L.; Luo, H.; Yang, Y.X.; Dai, X.Y.; Zhou, S.F.; et al. Upregulation of PD-L1 and APE1 is associated with tumorigenesis and poor prognosis of gastric cancer. Drug Des. Devel. Ther. 2015, 9, 901–909. [Google Scholar] [CrossRef]
- Sun, J.; Xu, K.; Wu, C.; Wang, Y.; Hu, Y.; Zhu, Y.; Chen, Y.; Shi, Q.; Yu, G.; Zhang, X. PD-L1 expression analysis in gastric carcinoma tissue and blocking of tumor-associated PD-L1 signaling by two functional monoclonal antibodies. Tissue Antigens 2007, 69, 19–27. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Chao, J.; Fuchs, C.S.; Shitara, K.; Tabernero, J.; Muro, K.; Van Cutsem, E.; Bang, Y.J.; De Vita, F.; Landers, G.; Yen, C.J.; et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol. 2021, 7, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Boku, N.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; Punt, C.J.; Hato, S.V.; Eleveld-Trancikova, D.; Jansen, B.J.; Nierkens, S.; Schreibelt, G.; de Boer, A.; Van Herpen, C.M.; Kaanders, J.H.; et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Investig. 2011, 121, 3100–3108. [Google Scholar] [CrossRef]
- Roselli, M.; Cereda, V.; di Bari, M.G.; Formica, V.; Spila, A.; Jochems, C.; Farsaci, B.; Donahue, R.; Gulley, J.L.; Schlom, J.; et al. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. Oncoimmunology 2013, 2, e27025. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Till, B.; Gao, Q. Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1331807. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Ajani, J.A.; Moehler, M.; Garrido, M.; Gallardo, C.; Shen, L.; Yamaguchi, K.; Wyrwicz, L.; Skoczylas, T.; Bragagnoli, A.C.; et al. Nivolumab plus chemotherapy or ipilimumab in gastro-oesophageal cancer. Nature 2022, 603, 942–948. [Google Scholar] [CrossRef]
- Kang, Y.K.; Chen, L.T.; Ryu, M.H.; Oh, D.Y.; Oh, S.C.; Chung, H.C.; Lee, K.W.; Omori, T.; Shitara, K.; Sakuramoto, S.; et al. Nivolumab plus chemotherapy versus placebo plus chemotherapy in patients with HER2-negative, untreated, unresectable advanced or recurrent gastric or gastro-oesophageal junction cancer (ATTRACTION-4): A randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022, 23, 234–247. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Nagasaka, T.; Kambara, T.; Hoshizima, N.; Murakami, J.; Sasamoto, H.; Hosokawa, M.; Naomoto, Y.; Isozaki, H.; Shimizu, K.; et al. Microsatellite instability and clinicopathological features in esophageal squamous cell cancer. Oncol. Rep. 2007, 18, 1123–1127. [Google Scholar]
- Campanella, N.C.; Lacerda, C.F.; Berardinelli, G.N.; Abrahao-Machado, L.F.; Cruvinel-Carloni, A.; De Oliveira, A.T.T.; Scapulatempo-Neto, C.; Crema, E.; Adad, S.J.; Rodrigues, M.A.M.; et al. Presence of microsatellite instability in esophageal squamous cell carcinoma associated with chagasic megaesophagus. Biomark Med. 2018, 12, 573–582. [Google Scholar] [CrossRef]
- Schulmann, K.; Brasch, F.E.; Kunstmann, E.; Engel, C.; Pagenstecher, C.; Vogelsang, H.; Kruger, S.; Vogel, T.; Knaebel, H.P.; Ruschoff, J.; et al. HNPCC-associated small bowel cancer: Clinical and molecular characteristics. Gastroenterology 2005, 128, 590–599. [Google Scholar] [CrossRef]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Oki, E.; Taniguchi, H.; Nakatani, K.; Aoki, D.; Kuwata, T.; Yoshino, T. The real-world data on microsatellite instability status in various unresectable or metastatic solid tumors. Cancer Sci. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Oh, D.Y.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Berton, D.; Curigliano, G.; Ellard, S.; Pérez, J.M.T.; Arkenau, H.-T.; Abdeddaim, C.; Moreno, V.; Guo, W.; Im, E.; et al. Safety and efficacy of anti–PD-1 antibody dostarlimab in patients (pts) with mismatch repair-deficient (dMMR) solid cancers: Results from GARNET study. J. Clin. Oncol. 2021, 39, 9. [Google Scholar] [CrossRef]
- Berton, D.; Banerjee, S.N.; Curigliano, G.; Cresta, S.; Arkenau, H.-T.; Abdeddaim, C.; Kristeleit, R.S.; Redondo, A.; Leath, C.A.; Torres, A.A.; et al. Antitumor activity of dostarlimab in patients with mismatch repair-deficient/microsatellite instability–high tumors: A combined analysis of two cohorts in the GARNET study. J. Clin. Oncol. 2021, 39, 2564. [Google Scholar] [CrossRef]
- Azad, N.S.; Gray, R.J.; Overman, M.J.; Schoenfeld, J.D.; Mitchell, E.P.; Zwiebel, J.A.; Sharon, E.; Streicher, H.; Li, S.; McShane, L.M.; et al. Nivolumab Is Effective in Mismatch Repair-Deficient Noncolorectal Cancers: Results From Arm Z1D-A Subprotocol of the NCI-MATCH (EAY131) Study. J. Clin. Oncol. 2020, 38, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Bergsland, E.; O’Neil, B.H.; Santoro, A.; Schellens, J.H.M.; Cohen, R.B.; Doi, T.; Ott, P.A.; Pishvaian, M.J.; Puzanov, I.; et al. Pembrolizumab for the treatment of programmed death-ligand 1-positive advanced carcinoid or pancreatic neuroendocrine tumors: Results from the KEYNOTE-028 study. Cancer 2020, 126, 3021–3030. [Google Scholar] [CrossRef]
- Strosberg, J.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.P.; Shapira-Frommer, R.; Bergsland, E.; Shah, M.; Fakih, M.; Takahashi, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Neuroendocrine Tumors: Results from the Phase II KEYNOTE-158 Study. Clin. Cancer Res. 2020, 26, 2124–2130. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, P.; Zhang, Y.; Li, Z.; Gong, J.; Li, J.; Li, J.; Li, Y.; Zhang, X.; Lu, Z.; et al. Efficacy, Safety, and Biomarkers of Toripalimab in Patients with Recurrent or Metastatic Neuroendocrine Neoplasms: A Multiple-Center Phase Ib Trial. Clin. Cancer Res. 2020, 26, 2337–2345. [Google Scholar] [CrossRef]
- Jia, R.; Li, Y.; Xu, N.; Jiang, H.P.; Zhao, C.H.; Liu, R.R.; Shi, Y.; Zhang, Y.Y.; Wang, S.Y.; Zhou, H.; et al. Sintilimab in Patients with Previously Treated Metastatic Neuroendocrine Neoplasms. Oncologist 2022, 27, e625–e632. [Google Scholar] [CrossRef]
- Klein, O.; Kee, D.; Markman, B.; Lee, R.C.; Michael, M.; Mileshkin, L.R.; Scott, C.L.; Linklater, R.; Menon, S.; Tebbutt, N.C.; et al. A phase II clinical trial of ipilimumab/nivolumab combination immunotherapy in patients with rare upper gastrointestinal, neuroendocrine, and gynecological malignancies. J. Clin. Oncol. 2019, 37, 2570. [Google Scholar] [CrossRef]
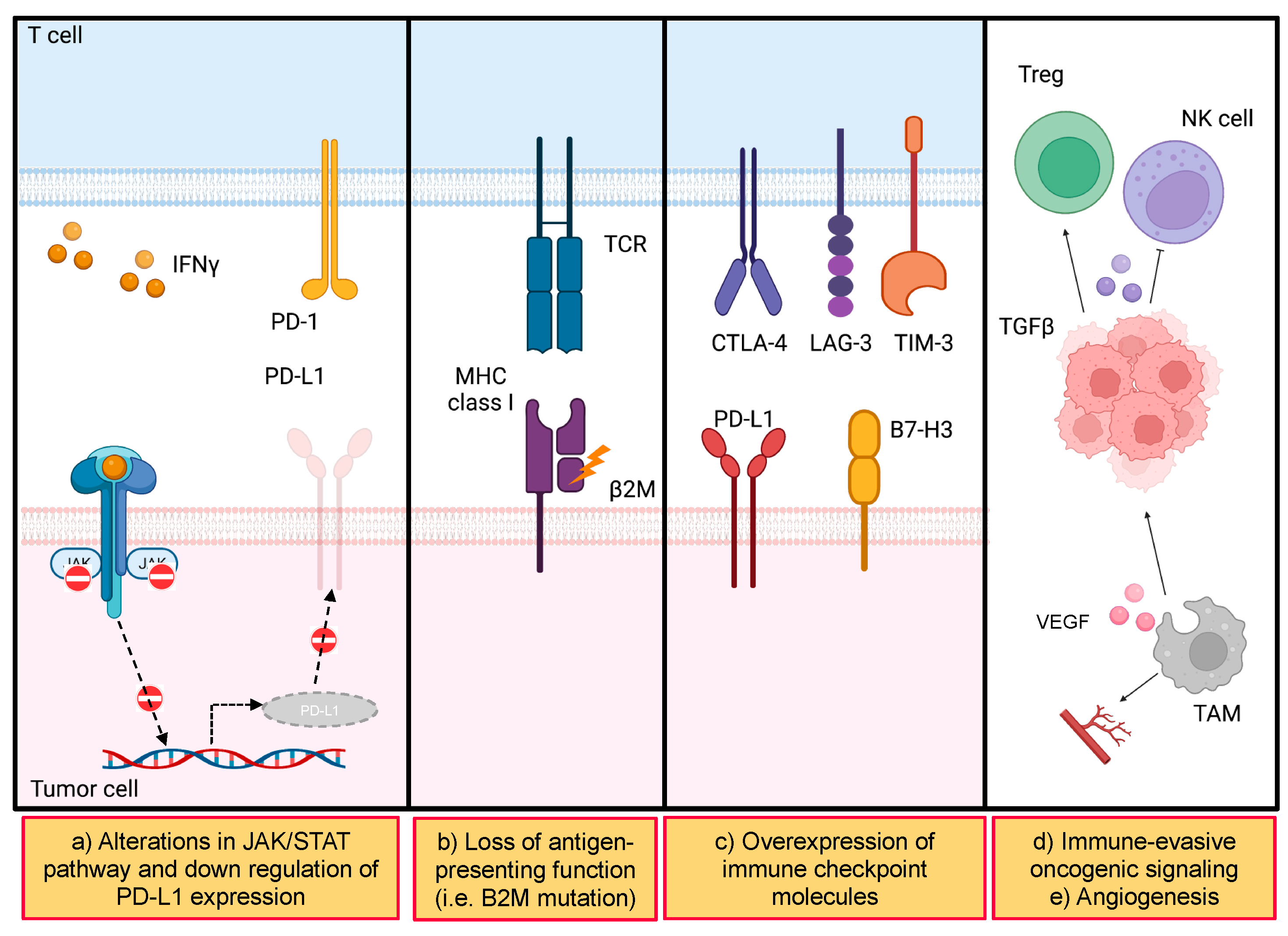
- Amodio, V.; Mauri, G.; Reilly, N.M.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A.; Germano, G. Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers 2021, 13, 2638. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Bortolomeazzi, M.; Keddar, M.R.; Montorsi, L.; Acha-Sagredo, A.; Benedetti, L.; Temelkovski, D.; Choi, S.; Petrov, N.; Todd, K.; Wai, P.; et al. Immunogenomics of Colorectal Cancer Response to Checkpoint Blockade: Analysis of the KEYNOTE 177 Trial and Validation Cohorts. Gastroenterology 2021, 161, 1179–1193. [Google Scholar] [CrossRef]
- Endo, E.; Okayama, H.; Saito, K.; Nakajima, S.; Yamada, L.; Ujiie, D.; Kase, K.; Fujita, S.; Endo, H.; Sakamoto, W.; et al. A TGFbeta-Dependent Stromal Subset Underlies Immune Checkpoint Inhibitor Efficacy in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Colorectal Cancer. Mol. Cancer Res. 2020, 18, 1402–1413. [Google Scholar] [CrossRef]
- Chida, K.; Kawazoe, A.; Kawazu, M.; Suzuki, T.; Nakamura, Y.; Nakatsura, T.; Kuwata, T.; Ueno, T.; Kuboki, Y.; Kotani, D.; et al. A Low Tumor Mutational Burden and PTEN Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/dMMR Gastrointestinal Tumors. Clin. Cancer Res. 2021, 27, 3714–3724. [Google Scholar] [CrossRef]
- Randrian, V.; Evrard, C.; Tougeron, D. Microsatellite Instability in Colorectal Cancers: Carcinogenesis, Neo-Antigens, Immuno-Resistance and Emerging Therapies. Cancers 2021, 13, 3063. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef]
- Abdullaev, S.; André, T.; Lei, M.; Lenz, H.-J.; Novotny, J.; Paulson, A.S.; Tejpar, S.; Yamazaki, K.; Ledeine, J.-M. A phase III study of nivolumab (NIVO), NIVO + ipilimumab (IPI), or chemotherapy (CT) for microsatellite instability-high (MSI-H)/mismatch repair-deficient (dMMR) metastatic colorectal cancer (mCRC): Checkmate 8HW. J. Clin. Oncol. 2020, 38, TPS266. [Google Scholar] [CrossRef]
- Kawakami, H.; Hironaka, S.; Esaki, T.; Chayama, K.; Tsuda, M.; Sugimoto, N.; Kadowaki, S.; Makiyama, A.; Machida, N.; Hirano, H.; et al. An Investigator-Initiated Phase 2 Study of Nivolumab Plus Low-Dose Ipilimumab as First-Line Therapy for Microsatellite Instability-High Advanced Gastric or Esophagogastric Junction Cancer (NO LIMIT, WJOG13320G/CA209-7W7). Cancers 2021, 13, 805. [Google Scholar] [CrossRef]
- Lima, C.M.S.P.R.; Yothers, G.; Jacobs, S.A.; Sanoff, H.K.; Cohen, D.J.; Guthrie, K.A.; Henry, N.L.; Ganz, P.A.; Kopetz, S.; Lucas, P.C.; et al. NRG-GI004/SWOG-S1610: Colorectal cancer metastatic dMMR immuno-therapy (COMMIT) study—A randomized phase III study of atezolizumab (atezo) monotherapy versus mFOLFOX6/bevacizumab/atezo in the first-line treatment of patients (pts) with deficient DNA mismatch repair (dMMR) or microsatellite instability high (MSI-H) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2022, 40, TPS232. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Lenz, H.J.; Ou, F.S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of Consensus Molecular Subtype on Survival in Patients With Metastatic Colorectal Cancer: Results From CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]
- de Miranda, N.F.; van Dinther, M.; van den Akker, B.E.; van Wezel, T.; ten Dijke, P.; Morreau, H. Transforming Growth Factor beta Signaling in Colorectal Cancer Cells with Microsatellite Instability Despite Biallelic Mutations in TGFBR2. Gastroenterology 2015, 148, 1427–1437.e8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pasche, B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16, R14–R20. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-beta Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef]
- Luo, J.; Chen, X.Q.; Li, P. The Role of TGF-beta and Its Receptors in Gastrointestinal Cancers. Transl. Oncol. 2019, 12, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, K.W.; Ahn, J.B.; Lee, J.; Ryu, J.; Oh, B.; Ock, C.-Y.; Hwang, S.; Hahm, K.B.; Kim, S.-J.; et al. Efficacy and safety of vactosertib and pembrolizumab combination in patients with previously treated microsatellite stable metastatic colorectal cancer. J. Clin. Oncol. 2021, 39, 3573. [Google Scholar] [CrossRef]
- Antoniotti, C.; Rossini, D.; Pietrantonio, F.; Catteau, A.; Salvatore, L.; Lonardi, S.; Boquet, I.; Tamberi, S.; Marmorino, F.; Moretto, R.; et al. Upfront FOLFOXIRI plus bevacizumab with or without atezolizumab in the treatment of patients with metastatic colorectal cancer (AtezoTRIBE): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2022, 23, 876–887. [Google Scholar] [CrossRef]
- Wallin, J.; Pishvaian, M.J.; Hernandez, G.; Yadav, M.; Jhunjhunwala, S.; Delamarre, L.; He, X.; Powderly, J.; Lieu, C.; Eckhardt, S.G.; et al. Abstract 2651: Clinical activity and immune correlates from a phase Ib study evaluating atezolizumab (anti-PDL1) in combination with FOLFOX and bevacizumab (anti-VEGF) in metastatic colorectal carcinoma. Cancer Res. 2016, 76, 2651. [Google Scholar] [CrossRef]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef]
- Pogue-Geile, K.; Yothers, G.; Taniyama, Y.; Tanaka, N.; Gavin, P.; Colangelo, L.; Blackmon, N.; Lipchik, C.; Kim, S.R.; Sharif, S.; et al. Defective mismatch repair and benefit from bevacizumab for colon cancer: Findings from NSABP C-08. J. Natl. Cancer Inst. 2013, 105, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Hoff, S.; Grünewald, S.; Röse, L.; Zopf, D. Immunomodulation by regorafenib alone and in combination with anti PD1 antibody on murine models of colorectal cancer. Ann. Oncol. 2017, 28, v423. [Google Scholar] [CrossRef]
- Tada, Y.; Togashi, Y.; Kotani, D.; Kuwata, T.; Sato, E.; Kawazoe, A.; Doi, T.; Wada, H.; Nishikawa, H.; Shitara, K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J. Immunother. Cancer 2018, 6, 106. [Google Scholar] [CrossRef]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef]
- Kawazoe, A.; Fukuoka, S.; Nakamura, Y.; Kuboki, Y.; Wakabayashi, M.; Nomura, S.; Mikamoto, Y.; Shima, H.; Fujishiro, N.; Higuchi, T.; et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.W.; Mao, L.; Yu, G.T.; Bu, L.L.; Ma, S.R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef]
- Garralda, E.; Sukari, A.; Lakhani, N.J.; Patnaik, A.; Lou, Y.; Im, S.-A.; Golan, T.; Geva, R.; Wermke, M.; Miguel, M.D.; et al. A phase 1 first-in-human study of the anti-LAG-3 antibody MK4280 (favezelimab) plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. J. Clin. Oncol. 2021, 39, 3584. [Google Scholar] [CrossRef]
- Hollebecque, A.; Chung, H.C.; de Miguel, M.J.; Italiano, A.; Machiels, J.P.; Lin, C.C.; Dhani, N.C.; Peeters, M.; Moreno, V.; Su, W.C.; et al. Safety and Antitumor Activity of alpha-PD-L1 Antibody as Monotherapy or in Combination with alpha-TIM-3 Antibody in Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Tumors. Clin. Cancer Res. 2021, 27, 6393–6404. [Google Scholar] [CrossRef]
- Bever, K.M.; Wang, H.; Durham, J.N.; Petrie, S.; Hoare, J.; Wilt, C.; Sharfman, W.H.; Azad, N.S.; Laheru, D.A.; Anders, R.A.; et al. Phase II study of nivolumab and relatlimab in advanced mismatch repair deficient (dMMR) cancers resistant to prior PD-(L)1 inhibition. J. Clin. Oncol. 2020, 38, TPS839. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; He, Y.; Ren, S.; Li, W.; Zhu, J.; Yu, J.; Wang, L.; Xiong, A.; Xu, N.; Mao, C.; et al. Phase Ia/Ib dose-escalation study of IBI110 (anti-LAG-3 mAb) as a single agent and in combination with sintilimab (anti-PD-1 mAb) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2021, 39, 2589. [Google Scholar] [CrossRef]
- Schoffski, P.; Tan, D.S.W.; Martin, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) +/− anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer 2022, 10, e003776. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Kanani, A.; Veen, T.; Soreide, K. Neoadjuvant immunotherapy in primary and metastatic colorectal cancer. Br. J. Surg. 2021, 108, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Verschoor, Y.L.; van den Berg, J.; Sikorska, K.; Beets, G.; Lent, A.V.; Grootscholten, M.C.; Aalbers, A.; Buller, N.; Marsman, H.; et al. LBA7 Neoadjuvant immune checkpoint inhibition in locally advanced MMR-deficient colon cancer: The NICHE-2 study. Ann. Oncol. 2022, 33, S1389. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- André, T.; Tougeron, D.; Piessen, G.; Fouchardière, C.d.l.; Louvet, C.; Adenis, A.; Jary, M.; Tournigand, C.; Aparicio, T.; Desrame, J.; et al. Neoadjuvant Nivolumab Plus Ipilimumab and Adjuvant Nivolumab in Localized Deficient Mismatch Repair/Microsatellite Instability-High Gastric or Esophagogastric Junction Adenocarcinoma: The GERCOR NEONIPIGA Phase II Study. J. Clin. Oncol. 2022, 41, 255–265. [Google Scholar] [CrossRef]
- Al-Batran, S.-E.; Lorenzen, S.; Thuss-Patience, P.C.; Homann, N.; Schenk, M.; Lindig, U.; Heuer, V.; Kretzschmar, A.; Goekkurt, E.; Haag, G.M.; et al. Surgical and pathological outcome, and pathological regression, in patients receiving perioperative atezolizumab in combination with FLOT chemotherapy versus FLOT alone for resectable esophagogastric adenocarcinoma: Interim results from DANTE, a randomized, multicenter, phase IIb trial of the FLOT-AIO German Gastric Cancer Group and Swiss SAKK. J. Clin. Oncol. 2022, 40, 4003. [Google Scholar] [CrossRef]
- Ludford, K.; Ho, W.J.; Thomas, J.V.; Raghav, K.P.S.; Murphy, M.B.; Fleming, N.D.; Lee, M.S.; Smaglo, B.G.; You, Y.N.; Tillman, M.M.; et al. Neoadjuvant Pembrolizumab in Localized Microsatellite Instability High/Deficient Mismatch Repair Solid Tumors. J. Clin. Oncol. 2023, JCO2201351. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, Y.L.; Berg, J.v.d.; Beets, G.; Sikorska, K.; Aalbers, A.; Lent, A.v.; Grootscholten, C.; Huibregtse, I.; Marsman, H.; Oosterling, S.; et al. Neoadjuvant nivolumab, ipilimumab, and celecoxib in MMR-proficient and MMR-deficient colon cancers: Final clinical analysis of the NICHE study. J. Clin. Oncol. 2022, 40, 3511. [Google Scholar] [CrossRef]
- Ludford, K.; Raghav, K.P.S.; Murphy, M.A.B.; Fleming, N.D.; Nelson, D.A.; Lee, M.S.; Smaglo, B.G.; You, Y.N.; Tillman, M.M.; Kamiya-Matsuoka, C.; et al. Safety and efficacy of neoadjuvant pembrolizumab in mismatch repair deficient localized/locally advanced solid tumors. J. Clin. Oncol. 2021, 39, 2520. [Google Scholar] [CrossRef]
- Bertagnolli, M.M.; Redston, M.; Compton, C.C.; Niedzwiecki, D.; Mayer, R.J.; Goldberg, R.M.; Colacchio, T.A.; Saltz, L.B.; Warren, R.S. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: Prospective evaluation of biomarkers for stages II and III colon cancer—A study of CALGB 9581 and 89803. J. Clin. Oncol. 2011, 29, 3153–3162. [Google Scholar] [CrossRef]
- Bando, H.; Tsukada, Y.; Inamori, K.; Togashi, Y.; Koyama, S.; Kotani, D.; Fukuoka, S.; Yuki, S.; Komatsu, Y.; Homma, S.; et al. Preoperative Chemoradiotherapy plus Nivolumab before Surgery in Patients with Microsatellite Stable and Microsatellite Instability-High Locally Advanced Rectal Cancer. Clin. Cancer Res. 2022, 28, 1136–1146. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Miceli, R.; Raimondi, A.; Kim, Y.W.; Kang, W.K.; Langley, R.E.; Choi, Y.Y.; Kim, K.M.; Nankivell, M.G.; Morano, F.; et al. Individual Patient Data Meta-Analysis of the Value of Microsatellite Instability As a Biomarker in Gastric Cancer. J. Clin. Oncol. 2019, 37, 3392–3400. [Google Scholar] [CrossRef]
- Sun, W.; Saeed, A.; Al-Rajabi, R.M.d.T.; Kasi, A.; Veeramachaneni, N.K.; Al-Kasspooles, M.M.; Baranda, J.C.; Phadnis, M.; Godwin, A.K.; Olyaee, M.; et al. A phase II study of perioperative mFOLFOX chemotherapy plus pembrolizumab combination in patients with potentially resectable adenocarcinoma of the esophageal, gastroesophageal junction (GEJ), and stomach. J. Clin. Oncol. 2022, 40, 329. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Cutsem, E.V.; Muro, K.; Wainberg, Z.A.; Al-Batran, S.-E.; Hyung, W.J.; Molena, D.; Evans, B.; Ruscica, D.; Robbins, S.H.; et al. MATTERHORN: Efficacy and safety of neoadjuvant-adjuvant durvalumab and FLOT chemotherapy in resectable gastric and gastroesophageal junction cancer—A randomized, double-blind, placebo-controlled, phase 3 study. J. Clin. Oncol. 2021, 39, TPS4151. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Fuchs, C.S.; Ohtsu, A.; Tabernero, J.; Ilson, D.H.; Hyung, W.J.; Strong, V.E.; Goetze, T.O.; Yoshikawa, T.; et al. KEYNOTE-585: Phase III study of perioperative chemotherapy with or without pembrolizumab for gastric cancer. Future Oncol. 2019, 15, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef]
- Stenzinger, A.; Allen, J.D.; Maas, J.; Stewart, M.D.; Merino, D.M.; Wempe, M.M.; Dietel, M. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 2019, 58, 578–588. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11, eaax7918. [Google Scholar] [CrossRef]
- Xiao, Y.; Freeman, G.J. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef]
- Shimozaki, K.; Hayashi, H.; Tanishima, S.; Horie, S.; Chida, A.; Tsugaru, K.; Togasaki, K.; Kawasaki, K.; Aimono, E.; Hirata, K.; et al. Concordance analysis of microsatellite instability status between polymerase chain reaction based testing and next generation sequencing for solid tumors. Sci. Rep. 2021, 11, 20003. [Google Scholar] [CrossRef]
- Middha, S.; Zhang, L.; Nafa, K.; Jayakumaran, G.; Wong, D.; Kim, H.R.; Sadowska, J.; Berger, M.F.; Delair, D.F.; Shia, J.; et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis Oncol. 2017, 2017, PO.17.00084. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Scroggins, S.M.; Hampel, H.L.; Turner, E.H.; Pritchard, C.C. Microsatellite instability detection by next generation sequencing. Clin. Chem. 2014, 60, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Okamoto, W.; Denda, T.; Nishina, T.; Komatsu, Y.; Yuki, S.; Yasui, H.; Esaki, T.; Sunakawa, Y.; Ueno, M.; et al. Clinical Validity of Plasma-Based Genotyping for Microsatellite Instability Assessment in Advanced GI Cancers: SCRUM-Japan GOZILA Substudy. JCO Precis Oncol. 2022, 6, e2100383. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef]
- Budczies, J.; Kluck, K.; Beck, S.; Ourailidis, I.; Allgauer, M.; Menzel, M.; Kazdal, D.; Perkhofer, L.; Kleger, A.; Schirmacher, P.; et al. Homologous recombination deficiency is inversely correlated with microsatellite instability and identifies immunologically cold tumors in most cancer types. J. Pathol. Clin. Res. 2022, 8, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]



| Study | Patient Population | No. of Patients with MSI-H/dMMR Solid Tumors | MSI-H/dMMR Testing | Dose | Prior Therapy |
|---|---|---|---|---|---|
| KEYNOTE-016 [6] NCT01867511 | CRC and other tumors | 28 CRC 30 non-CRC | PCR or IHC prospectively evaluated | 10 mg/kg every 2 weeks | CRC: ≥2 prior regimens Non-CRC: ≥1 prior regimen |
| KEYNOTE-164 [21] NCT02460198 | CRC | 61 | PCR or IHC prospectively evaluated | 200 mg every 3 weeks | Prior fluoropyrimidine, oxaliplatin, and irinotecan +/− anti-VEGF/EGFR mAb |
| KEYNOTE-012 [29] NCT01848834 | PD-L1–positive gastric, bladder, or triple-negative breast cancer | 6 | PCR retrospectively identified | 10 mg/kg every 2 weeks | ≥1 prior regimen |
| KEYNOTE-028 [28] NCT02054806 | PD-L1–positive esophageal, biliary, breast, endometrial, or CRC | 5 | PCR retrospectively identified | 10 mg/kg every 2 weeks | ≥1 prior regimen |
| KEYNOTE-158 [27] NCT02628067 | MSI-H/dMMR non-CRC | 19 | PCR or IHC prospectively evaluated (retrospectively identified in specific rare tumors) | 200 mg every 3 weeks | ≥1 prior regimen |
| Study | KEYNOTE-059 [42] | KEYNOTE-061 [42] | KEYNOTE-062 [42] | CheckMate-649 [53] | |||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment line | ≥3rd line | 2nd line | 1st line | 1st line | |||||
| Phase | II | III | III | III | |||||
| No. of MSI-H/dMMR patients | 7 | 15 | 12 | 14 CPS ≥ 1 | 17 CPS ≥ 1 | 19 CPS ≥ 1 | 23 | 11 | 21 |
| Primary endpoint | OS and PFS in PD-L1CPS ≥ 1 | OS and PFS in PD-L1CPS ≥ 1 | PS and PFS in PD-L1 CPS ≥ 5 | ||||||
| Regimen | Pembro | Pembro | Chemo | Pembro | Chemo + Pembro | Chemo | Chemo + Nivo | Nivo1 + Ipi3 | Chemo |
| ORR | 57.1% | 46.7% | 16.7% | 57.1% | 64.7% | 36.8% | 55% | 70% | 39% |
| DoR | NR | NR | NR | 21.2 | NR | 7.0 | − | − | − |
| Median PFS | NR | 17.8 | 3.5 | 11.2 | NR | 6.6 | − | − | − |
| HR for PFS (95% CI) | − | − | − | 0.72 (0.31–1.68) | 0.45 (0.18–1.11) | Ref | − | − | − |
| 12-month PFS rate | − | − | − | 43% | 56% | 28% | − | − | − |
| Median OS | NR | NR | 8.1 | NR | NR | 8.5 | 38.7 | NR | 12.3 |
| HR for OS (95% CI) | − | 0.42 (0.13–1.31) | Ref | 0.21 (0.06–0.83) | 0.37 (0.14–0.97) | Ref | 0.38 (0.17–0.84) | 0.28 (0.08–0.92) | Ref |
| 12-month OS rate | 71% | 73% | 25% | 79% | 71% | 47% | − | − | − |
| Study | NICHE-2 [116,117] | Cercek et al. [118] | GERCOR NEONIPIGA [119] | DANTE [120] | Kaysia et al. [121] |
|---|---|---|---|---|---|
| NCT number | NCT03026140 | NCT04165772 | NCT04006262 | NCT03421288 | NCT04082572 |
| Primary tumor | Colorectal | Rectal | Gastric or gastroesophageal junction cancer | Gastric or gastroesophageal cancer | Solid tumors |
| Phase | Phase II | Phase II | Phase II | Randomized phase II | Phase II |
| Number of patients with MSI-H/dMMR tumor | 112 | 12 (ongoing) | 32 | 8 (8 patients with FLOT plus atezolizumab arm) | 35 |
| Clinical stage | I–III | II/III | cT2–4, any N | cT2–4, any N | Unresectable or high-risk resectable |
| Regimen | Nivolumab plus ipilimumab | Dostarlimab | Nivolumab plus ipilimumab | FLOT plus atezolizumab | Pembrolizumab |
| Primary endpoints |
|
|
|
|
|
| Clinical CR (95% CI) | Not reported | 100% (74–100) | NA | NA | 30% |
| Pathological CR | 67% | NA | 58.6% | 63% | 65% (79% in CRC) |
| MPR | 95% | NA | 72.4% | 75% | NA |
| Adverse events of grade ≥3 | 13% | 0% | 25% | Not reported | 6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimozaki, K.; Nakayama, I.; Hirota, T.; Yamaguchi, K. Current Strategy to Treat Immunogenic Gastrointestinal Cancers: Perspectives for a New Era. Cells 2023, 12, 1049. https://doi.org/10.3390/cells12071049
Shimozaki K, Nakayama I, Hirota T, Yamaguchi K. Current Strategy to Treat Immunogenic Gastrointestinal Cancers: Perspectives for a New Era. Cells. 2023; 12(7):1049. https://doi.org/10.3390/cells12071049
Chicago/Turabian StyleShimozaki, Keitaro, Izuma Nakayama, Toru Hirota, and Kensei Yamaguchi. 2023. "Current Strategy to Treat Immunogenic Gastrointestinal Cancers: Perspectives for a New Era" Cells 12, no. 7: 1049. https://doi.org/10.3390/cells12071049
APA StyleShimozaki, K., Nakayama, I., Hirota, T., & Yamaguchi, K. (2023). Current Strategy to Treat Immunogenic Gastrointestinal Cancers: Perspectives for a New Era. Cells, 12(7), 1049. https://doi.org/10.3390/cells12071049