

Tumor Microenvironmental Cytokines Drive NSCLC Cell Aggressiveness and Drug-Resistance via YAP-Mediated Autophagy
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture Treatments
2.2. Cell Migration Assay
2.3. Cell Invasion Assay
2.4. Immunoblot Analysis
2.5. Fluorescence Microscopy
2.6. siRNAs Transfection
2.7. Flow Cytometry Analyses
2.8. Data Analysis and Statistics
3. Results
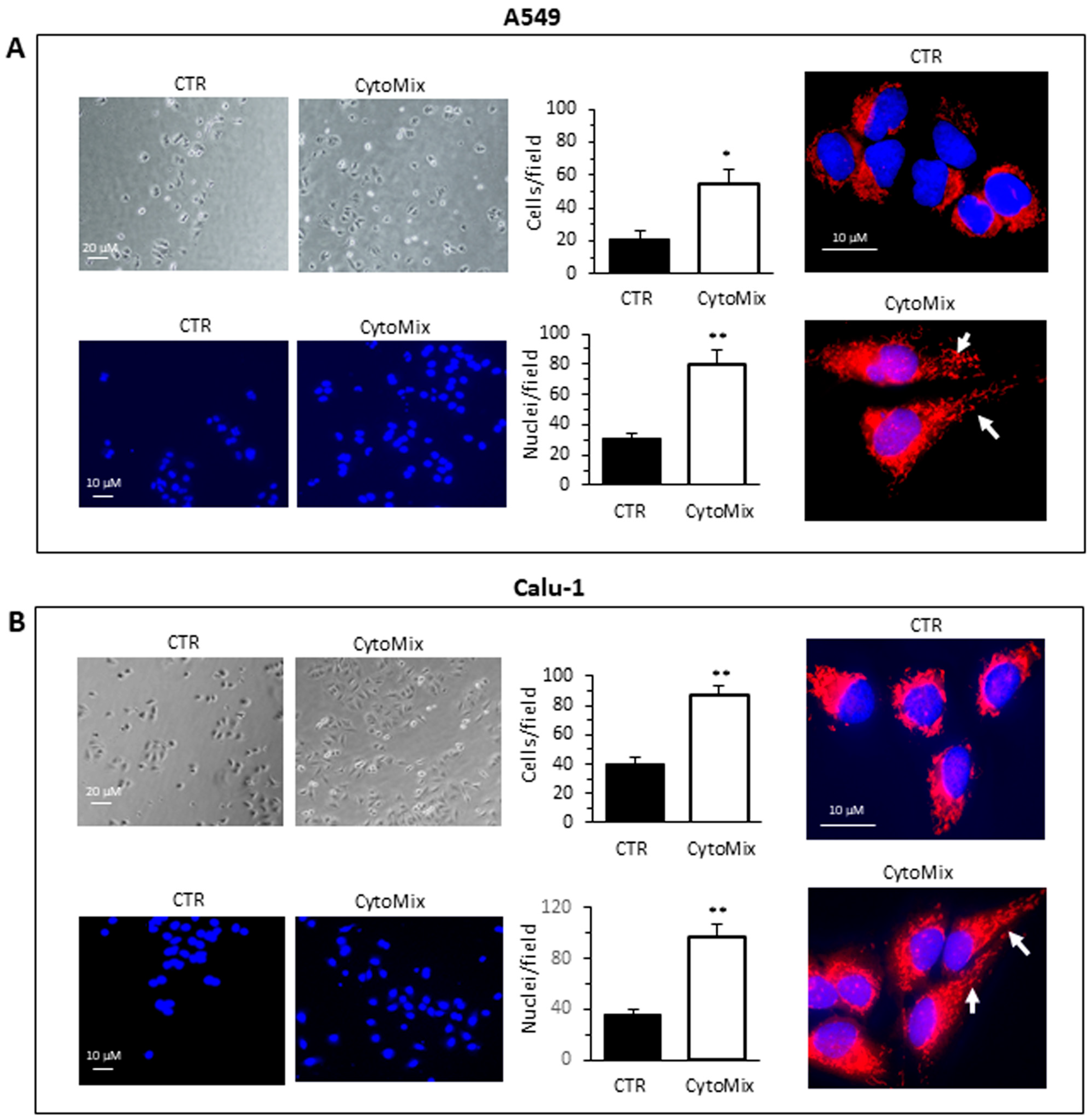
3.1. CytoMix Promotes Cell Motility and Invasiveness in NSCLC Cells
3.2. CytoMix Promotes Epithelial–Mesenchymal Transition (EMT) and Increases Cell Surface Expression of Stem Cell Markers and GSH Content in NSCLC Cells
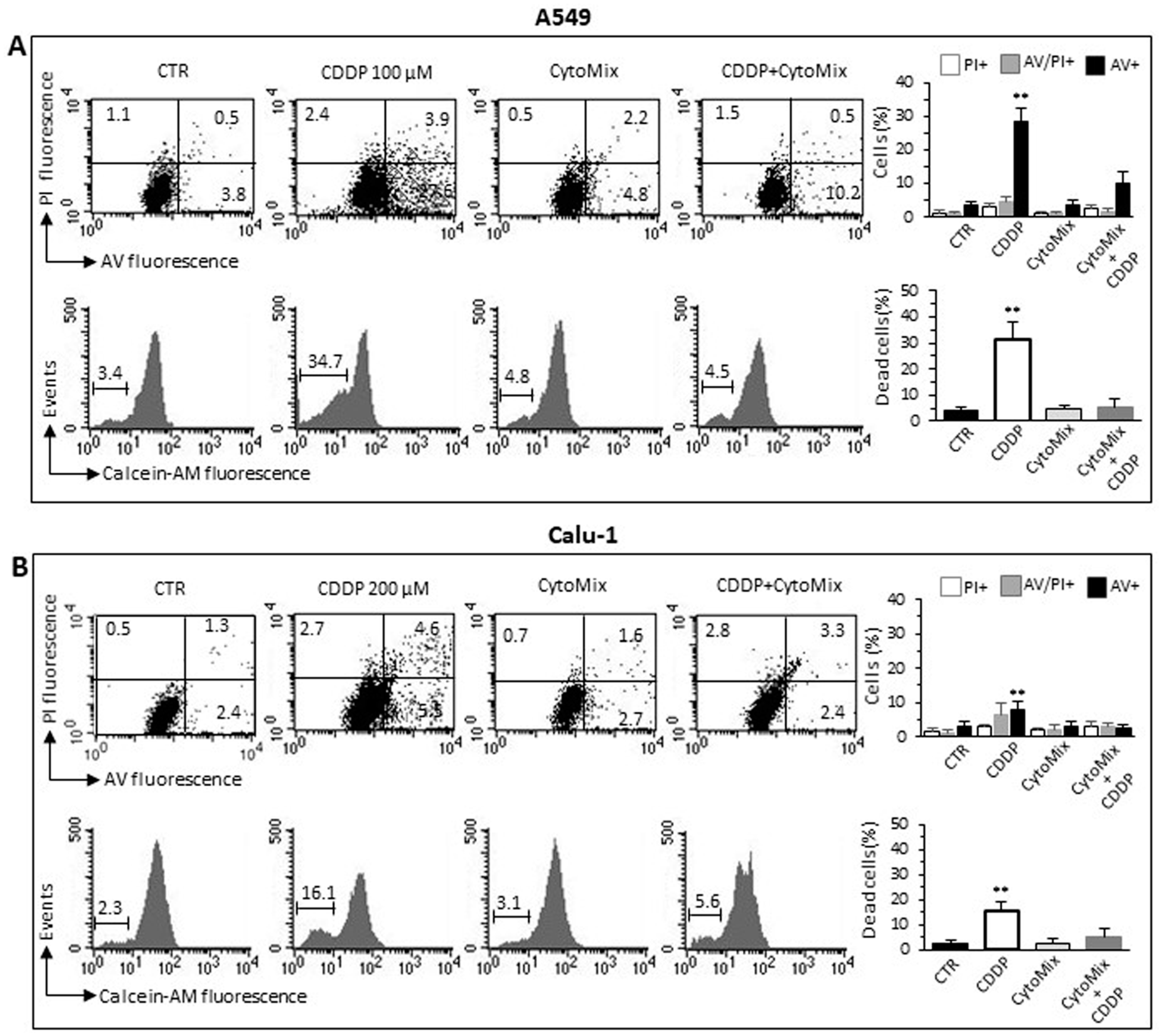
3.3. CytoMix Reduces the CDDP Toxicity in NSCLC Cells
3.4. CytoMix Induces Autophagy Process via YAP Signaling in NSCLC Cells
3.5. YAP siRNA Inhibits CytoMix-Induced Cell Autophagy and Invasion in NSCLC Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Altorki, N.K.; Borczuk, A.C.; Harrison, S.; Groner, L.K.; Bhinder, B.; Mittal, V.; Elemento, O.; McGraw, T.E. Global evolution of the tumor microenvironment associated with progression from preinvasive invasive to invasive human lung adenocarcinoma. Cell Rep. 2022, 39, 110639. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol. Rev. 2021, 302, 259–272. [Google Scholar] [CrossRef]
- Hu, H.; Piotrowska, Z.; Hare, P.J.; Chen, H.; Mulvey, H.E.; Mayfield, A.; Noeen, S.; Kattermann, K.; Greenberg, M.; Williams, A.; et al. Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell 2021, 39, 1531–1547.e10. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Melchionna, R.; Spada, S.; Di Modugno, F.; D’Andrea, D.; Di Carlo, A.; Panetta, M.; Mileo, A.M.; Sperduti, I.; Antoniani, B.; Gallo, E.; et al. The actin modulator hMENA regulates GAS6-AXL axis and pro-tumor cancer/stromal cell cooperation. EMBO Rep. 2020, 21, e50078. [Google Scholar] [CrossRef]
- Linares, J.; Marin-Jimenez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and Functions of CAFs Secretome During Cancer Progression and Therapy. Front. Cell Dev. Biol. 2020, 8, 621070. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Cytoprotective and nonprotective autophagy in cancer therapy. Autophagy 2013, 9, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-mediated tumor promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24. [Google Scholar] [CrossRef] [PubMed]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Dupont, N.; Castillo, E.F.; Deretic, V. Secretory versus degradative autophagy: Unconventional secretion of inflammatory mediators. J. Innate Immun. 2013, 5, 471–479. [Google Scholar] [CrossRef]
- New, J.; Thomas, S.M. Autophagy-dependent secretion: Mechanism, factors secreted, and disease implications. Autophagy 2019, 15, 1682–1693. [Google Scholar] [CrossRef]
- Soreng, K.; Neufeld, T.P.; Simonsen, A. Membrane Trafficking in Autophagy. Int. Rev. Cell Mol. Biol. 2018, 336, 1–92. [Google Scholar] [CrossRef]
- Raudenska, M.; Balvan, J.; Masarik, M. Crosstalk between autophagy inhibitors and endosome-related secretory pathways: A challenge for autophagy-based treatment of solid cancers. Mol. Cancer 2021, 20, 140. [Google Scholar] [CrossRef]
- Wang, D.; He, J.; Huang, B.; Liu, S.; Zhu, H.; Xu, T. Emerging role of the Hippo pathway in autophagy. Cell Death Dis. 2020, 11, 880. [Google Scholar] [CrossRef]
- Totaro, A.; Zhuang, Q.; Panciera, T.; Battilana, G.; Azzolin, L.; Brumana, G.; Gandin, A.; Brusatin, G.; Cordenonsi, M.; Piccolo, S. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc. Natl. Acad. Sci. USA 2019, 116, 17848–17857. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Li, F.L.; Guan, K.L. The two sides of Hippo pathway in cancer. Semin. Cancer Biol. 2022, 85, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Jiang, J. Hippo-Independent Regulation of Yki/Yap/Taz: A Non-canonical View. Front. Cell Dev. Biol. 2021, 9, 658481. [Google Scholar] [CrossRef]
- Pavel, M.; Renna, M.; Park, S.J.; Menzies, F.M.; Ricketts, T.; Fullgrabe, J.; Ashkenazi, A.; Frake, R.A.; Lombarte, A.C.; Bento, C.F.; et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat. Commun. 2018, 9, 2961. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Matarrese, P.; Abbruzzese, C.; Mileo, A.M.; Vona, R.; Ascione, B.; Visca, P.; Rollo, F.; Benevolo, M.; Malorni, W.; Paggi, M.G. Interaction between the human papillomavirus 16 E7 oncoprotein and gelsolin ignites cancer cell motility and invasiveness. Oncotarget 2016, 7, 50972–50985. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Sebastia, J.; Cristofol, R.; Martin, M.; Rodriguez-Farre, E.; Sanfeliu, C. Evaluation of fluorescent dyes for measuring intracellular glutathione content in primary cultures of human neurons and neuroblastoma SH-SY5Y. Cytom. A 2003, 51, 16–25. [Google Scholar] [CrossRef]
- Riemann, D.; Tcherkes, A.; Hansen, G.H.; Wulfaenger, J.; Blosz, T.; Danielsen, E.M. Functional co-localization of monocytic aminopeptidase N/CD13 with the Fc gamma receptors CD32 and CD64. Biochem. Biophys. Res. Commun. 2005, 331, 1408–1412. [Google Scholar] [CrossRef]
- Kondoh, N.; Mizuno-Kamiya, M. The Role of Immune Modulatory Cytokines in the Tumor Microenvironments of Head and Neck Squamous Cell Carcinomas. Cancers 2022, 14, 2884. [Google Scholar] [CrossRef]
- Shi, X.; Luo, J.; Weigel, K.J.; Hall, S.C.; Du, D.; Wu, F.; Rudolph, M.C.; Zhou, H.; Young, C.D.; Wang, X.J. Cancer-Associated Fibroblasts Facilitate Squamous Cell Carcinoma Lung Metastasis in Mice by Providing TGFbeta-Mediated Cancer Stem Cell Niche. Front. Cell Dev. Biol. 2021, 9, 668164. [Google Scholar] [CrossRef]
- Hu, W.; Liu, Y.; Zhou, W.; Si, L.; Ren, L. CXCL16 and CXCR6 are coexpressed in human lung cancer in vivo and mediate the invasion of lung cancer cell lines in vitro. PLoS ONE 2014, 9, e99056. [Google Scholar] [CrossRef]
- Vona, R.; Mileo, A.M.; Matarrese, P. Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers 2021, 13, 5812. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Herreros-Pomares, A.; de-Maya-Girones, J.D.; Calabuig-Farinas, S.; Lucas, R.; Martinez, A.; Pardo-Sanchez, J.M.; Alonso, S.; Blasco, A.; Guijarro, R.; Martorell, M.; et al. Lung tumorspheres reveal cancer stem cell-like properties and a score with prognostic impact in resected non-small-cell lung cancer. Cell Death Dis. 2019, 10, 660. [Google Scholar] [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nistico, P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Kang, Y.; Li, X.; Ma, X.; Ma, L. MCAM expression is associated with poor prognosis in non-small cell lung cancer. Clin. Transl. Oncol. 2014, 16, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Osude, C.; Lin, L.; Patel, M.; Eckburg, A.; Berei, J.; Kuckovic, A.; Dube, N.; Rastogi, A.; Gautam, S.; Smith, T.J.; et al. Mediating EGFR-TKI Resistance by VEGF/VEGFR Autocrine Pathway in Non-Small Cell Lung Cancer. Cells 2022, 11, 1694. [Google Scholar] [CrossRef] [PubMed]
- Liping, Y.; Jian, H.; Zhenchao, T.; Yan, Z.; Jing, Y.; Yangyang, Z.; Jing, G.; Liting, Q. GSH-responsive poly-resveratrol based nanoparticles for effective drug delivery and reversing multidrug resistance. Drug Deliv. 2022, 29, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, Y.; Duan, W.; Song, J.; Wei, S.; Xia, S.; Wang, Y.; Du, X.; Li, E.; Ren, C.; et al. Glutathione peroxidase 4-dependent glutathione high-consumption drives acquired platinum chemoresistance in lung cancer-derived brain metastasis. Clin. Transl. Med. 2021, 11, e517. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Choi, N.; Kim, K.; Koo, H.J.; Choi, J.; Kim, H.N. Chemoresistance of Cancer Cells: Requirements of Tumor Microenvironment-mimicking In Vitro Models in Anti-Cancer Drug Development. Theranostics 2018, 8, 5259–5275. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, X.; Ren, Y.; Geng, H.; Zhang, Q.; Cao, L.; Meng, Z.; Wu, X.; Xu, M.; Xu, K. Cancer-associated fibroblasts contribute to cisplatin resistance by modulating ANXA3 in lung cancer cells. Cancer Sci. 2019, 110, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef]
- Barlesi, F.; Garon, E.B.; Kim, D.W.; Felip, E.; Han, J.Y.; Kim, J.H.; Ahn, M.J.; Fidler, M.J.; Gubens, M.A.; de Castro, G., Jr.; et al. Health-Related Quality of Life in KEYNOTE-010: A Phase II/III Study of Pembrolizumab Versus Docetaxel in Patients With Previously Treated Advanced, Programmed Death Ligand 1-Expressing NSCLC. J. Thorac. Oncol. 2019, 14, 793–801. [Google Scholar] [CrossRef]
- Rossi, A.; Di Maio, M. Platinum-based chemotherapy in advanced non-small-cell lung cancer: Optimal number of treatment cycles. Expert Rev. Anticancer Ther. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Christofori, G. The cross-talk between the Hippo signaling pathway and autophagy:implications on physiology and cancer. Cell Cycle 2020, 19, 2563–2572. [Google Scholar] [CrossRef]
- Lee, Y.A.; Noon, L.A.; Akat, K.M.; Ybanez, M.D.; Lee, T.F.; Berres, M.L.; Fujiwara, N.; Goossens, N.; Chou, H.I.; Parvin-Nejad, F.P.; et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat. Commun. 2018, 9, 4962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, F.; Chen, S.; Plouffe, S.W.; Wu, S.; Liu, S.; Li, X.; Zhou, R.; Wang, J.; Zhao, B.; et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 2017, 19, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Sahai, E.; Rullan, A. SnapShot: Cancer-Associated Fibroblasts. Cell 2020, 181, 486–486.e1. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Ringuette Goulet, C.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFbeta Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef]
- Hu, T.; Hu, J. Melanoma-derived exosomes induce reprogramming fibroblasts into cancer-associated fibroblasts via Gm26809 delivery. Cell Cycle 2019, 18, 3085–3094. [Google Scholar] [CrossRef]
- Dror, S.; Sander, L.; Schwartz, H.; Sheinboim, D.; Barzilai, A.; Dishon, Y.; Apcher, S.; Golan, T.; Greenberger, S.; Barshack, I.; et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 2016, 18, 1006–1017. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Ngabire, D.; Kim, G.D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Altieri, D.C. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin. Cancer Res. 2016, 22, 540–545. [Google Scholar] [CrossRef]
- Schuler, M.H.; Lewandowska, A.; Caprio, G.D.; Skillern, W.; Upadhyayula, S.; Kirchhausen, T.; Shaw, J.M.; Cunniff, B. Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 2017, 28, 2159–2169. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, J.; Wang, X.; Wei, N.; Liu, H.; Zhang, X. CD146-mediated acquisition of stemness phenotype enhances tumour invasion and metastasis after EGFR-TKI resistance in lung cancer. Clin. Respir. J. 2019, 13, 23–33. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.Y.; Jang, G.B.; Choi, J.H.; Kim, J.H.; Lee, C.J.; Lee, S.; Baek, J.H.; Park, K.K.; Kim, J.M.; et al. Aberrant activation of the CD45-Wnt signaling axis promotes stemness and therapy resistance in colorectal cancer cells. Theranostics 2021, 11, 8755–8770. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, H.; Liao, Y.; Wu, W.; Liu, L.; Liu, L.; Wu, Y.; Sun, F.; Lin, H.W. Inhibition of Wnt/beta-catenin pathway reverses multi-drug resistance and EMT in Oct4(+)/Nanog(+) NSCLC cells. Biomed. Pharmacother. 2020, 127, 110225. [Google Scholar] [CrossRef]
- Bhardwaj, V.; He, J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int. J. Mol. Sci. 2020, 21, 3412. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Jang, M.; Song, M.J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Chen, B.; Jiao, N.L.; Liu, Y.H.; Sun, S.Y.; Zhang, Y.W. Elevated Glutathione Peroxidase 2 Expression Promotes Cisplatin Resistance in Lung Adenocarcinoma. Oxid. Med. Cell. Longev. 2020, 2020, 7370157. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, X.; Fu, G.; Ge, M. Glutathione peroxidase 2 overexpression promotes malignant progression and cisplatin resistance of KRAS-mutated lung cancer cells. Oncol. Rep. 2022, 48, 207. [Google Scholar] [CrossRef]
- Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers 2021, 13, 5622. [Google Scholar] [CrossRef]
- Maheswari, U.; Sadras, S.R. Mechanism and Regulation of Autophagy in Cancer. Crit. Rev. Oncog. 2018, 23, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Camuzard, O.; Santucci-Darmanin, S.; Carle, G.F.; Pierrefite-Carle, V. Autophagy in the crosstalk between tumor and microenvironment. Cancer Lett. 2020, 490, 143–153. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef]
- Xu, M.; Gu, M.; Zhou, J.; Da, J.; Wang, Z. Interaction of YAP1 and mTOR promotes bladder cancer progression. Int. J. Oncol. 2020, 56, 232–242. [Google Scholar] [CrossRef]
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Kumar, S.; Nishimura, R.N.; Gera, J. mTORC2-mediated direct phosphorylation regulates YAP activity promoting glioblastoma growth and invasive characteristics. Neoplasia 2021, 23, 951–965. [Google Scholar] [CrossRef]
- Frasa, M.A.; Maximiano, F.C.; Smolarczyk, K.; Francis, R.E.; Betson, M.E.; Lozano, E.; Goldenring, J.; Seabra, M.C.; Rak, A.; Ahmadian, M.R.; et al. Armus is a Rac1 effector that inactivates Rab7 and regulates E-cadherin degradation. Curr. Biol. 2010, 20, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Mack, N.A.; Whalley, H.J.; Castillo-Lluva, S.; Malliri, A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011, 10, 1571–1581. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matarrese, P.; Vona, R.; Ascione, B.; Cittadini, C.; Tocci, A.; Mileo, A.M. Tumor Microenvironmental Cytokines Drive NSCLC Cell Aggressiveness and Drug-Resistance via YAP-Mediated Autophagy. Cells 2023, 12, 1048. https://doi.org/10.3390/cells12071048
Matarrese P, Vona R, Ascione B, Cittadini C, Tocci A, Mileo AM. Tumor Microenvironmental Cytokines Drive NSCLC Cell Aggressiveness and Drug-Resistance via YAP-Mediated Autophagy. Cells. 2023; 12(7):1048. https://doi.org/10.3390/cells12071048
Chicago/Turabian StyleMatarrese, Paola, Rosa Vona, Barbara Ascione, Camilla Cittadini, Annalisa Tocci, and Anna Maria Mileo. 2023. "Tumor Microenvironmental Cytokines Drive NSCLC Cell Aggressiveness and Drug-Resistance via YAP-Mediated Autophagy" Cells 12, no. 7: 1048. https://doi.org/10.3390/cells12071048
APA StyleMatarrese, P., Vona, R., Ascione, B., Cittadini, C., Tocci, A., & Mileo, A. M. (2023). Tumor Microenvironmental Cytokines Drive NSCLC Cell Aggressiveness and Drug-Resistance via YAP-Mediated Autophagy. Cells, 12(7), 1048. https://doi.org/10.3390/cells12071048